| Startseite |
Im MO-Modell wird die Elektronenstruktur eines Moleküls durch delokalisierte
Molekülorbitale beschrieben. Diese MO's werden durch Linearkombination der
Atomorbitale gebildet.
Im VB-Modell wird die Elektronenstruktur durch Geminale beschrieben. Diese
werden ebenfalls aus Atomorbitalen gebildet, allerdings aus jeweils nur zwei
AO's, und zwar aus den zwei AO's, die eine Bindung beschreiben. Diese beiden
Bindungsorbitale enthalten je ein Elektron, die miteinander "koppeln", d. h. ein
Elektronenpaar bilden, welches die Bindung (Valenz) darstellt. Das Geminal
stellt also eine Zwei-Elektronen-Wellenfunktion dar (Gemini = Zwillinge).
Im Folgenden soll an einigen Beispielen eine GVB-Berechnung (Reference) diskutiert werden. Wir beginnen mit dem Ozon-Molekül, da man die Elektronenstruktur dieses Moleküls häufig im VB-Bild diskutiert.
Inputfile
!
! O3-Molekuel, GVB, Koordinaten: experimentell
!
$CONTRL SCFTYP=GVB MULT=1 RUNTYP=ENERGY
COORD=CART $END
$SYSTEM TIMLIM=1000 MEMORY=5000000 $END
$SCF NCO=12 NSETO=0 NPAIR=0 NO(1)=0 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
O3-Molekuel, GVB
CNV 2
O1 8.0 0.0000 0.0000 0.0000
O2 8.0 0.0000 1.0885 0.6697
O3 8.0 0.0000 -1.0885 0.6697
$END
NCO =12: Zahl der abgeschlossenen
Schalen. Wir benutzen 12 Elektronenpaare (= 24 Elektronen)
NSETO =0: The number of sets of open
shells
in the function. Maximum of
10. Keine ungepaarten Elektronen.
NPAIR =0: The number of geminal
pairs in the -GVB- function. Maximum of
12. The default corresponds to open shell SCF (default=0).
NO(1) =0: An array giving the degeneracy of each open shell set. Give NSETO
values. (default=0,0,0,...).
NPAIR=0 entspricht einer Valenzstruktur mit
abgeschlossenen Schalen, d. h. mit nur einer Grenzstruktur. Die Berechnung
sollte identisch sein mit einer RHF-Rechnung. Die Resonanz zweier
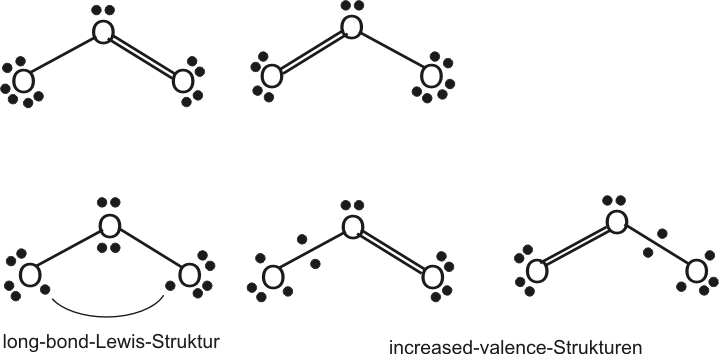
Valenzstrukturen (folgende Abbildung, oben) würde man durch NPAIR=1 ausdrücken.
Es liegt ein "aktives" spin-gepaartes Elektronenpaar vor.
Die erste untere Struktur (long-bond) entspricht einem "spin-ungepaarten", biradikalischen Elektronenpaar, die beiden anderen (increased-valence) zwei ungepaarten Elektronenpaaren.
Output
*************************
ROHF-GVB INPUT PARAMETERS
*************************
NORB = 12 NCO = 12
NPAIR = 0
NSETO = 0
----------------------------
ROHF-GVB COUPLING PARAMETERS
----------------------------
F VECTOR (OCCUPANCIES)
1 1.0000000000
ALPHA COUPLING COEFFICEINTS
1
1 2.0000000
BETA COUPLING COEFFICIENTS
1
1 -1.0000000
mit
$SCF NCO=11 NSETO=0 NPAIR=1
NO=0 $END
d.h. Grenzstrukturen mit einem aktiven Elektronenpaar (1.Grenzstruktur der
zweiten Zeile in der obigen Abbildung).
*************************
ROHF-GVB INPUT PARAMETERS
*************************
NORB = 13 NCO = 11
NPAIR = 1 NSETO = 0
PAIR ORBITALS
PAIR 1 HAS ORBS 12 13
Man sieht, dass das eine aktive
Elektronenpaar die Zahl der Geminale um Eins erhöht: NORB=13. Es wird ein
virtuelles Orbital zur Beschreibung der Elektronenstruktur benutzt.
----------------------------
ROHF-GVB COUPLING PARAMETERS
----------------------------
F VECTOR (OCCUPANCIES)
1 1.0000000000
2 0.9529411765
3 0.0470588235
Diese Angabe suggeriert, dass auch
noch ein zweites virtuelles Orbital schwach besetzt ist (0.047)
ALPHA COUPLING COEFFICEINTS
1 2 3
1 2.0000000
2 1.9058824 0.9529412
3 0.0941176 0.0000000 0.0470588
BETA COUPLING COEFFICIENTS
1 2 3
1 -1.0000000
2 -0.9529412 0.0000000
3 -0.0470588 -0.2117647 0.0000000
NATURAL ORBITAL COEFFICIENTS
N.O. PAIR CICOEF-S
1 0.9761870602 -0.2169304578
mit
$SCF NCO=11 NSETO=2 NO(1)=1,1 $END
eine biradikalische Grenzstruktur?
EXTRAPOLATION IN EFFECT
SOSCF IN EFFECT
*************************
ROHF-GVB INPUT PARAMETERS
*************************
NORB = 13 NCO = 11
NPAIR = 0 NSETO = 2
NO = 1 1
OPEN SHELL ORBITALS
SET 1 HAS ORBS 12
SET 2 HAS ORBS 13
----------------------------
ROHF-GVB COUPLING PARAMETERS
----------------------------
F VECTOR (OCCUPANCIES)
1 1.0000000000
2 0.5000000000
3 0.5000000000
ALPHA COUPLING COEFFICEINTS
1 2 3
1 2.0000000
2 1.0000000 0.0000000
3 1.0000000 0.5000000 0.0000000
BETA COUPLING COEFFICIENTS
1 2 3
1 -1.0000000
2 -0.5000000 0.0000000
3 -0.5000000 0.5000000 0.0000000
mit
$SCF NCO=10 NPAIR=1 NSETO=2 NO(1)=1,1 $END
zwei aktive Grenzstrukturen: eine
resonante Bindung, eine biradikalische?
*************************
ROHF-GVB INPUT PARAMETERS
*************************
NORB = 14 NCO = 10
NPAIR = 1 NSETO = 2
NO = 1 1
OPEN SHELL ORBITALS
SET 1 HAS ORBS 11
SET 2 HAS ORBS 12
PAIR ORBITALS
PAIR 1 HAS ORBS 13 14
Jetzt wird die Sache etwas
deutlicher. Von den ursprünglich 12 doppelt besetzten Orbitalen werden 10
"eingefroren" (NCO=10). Zusätzlich werden zwei virtuelle Orbitale benutzt, so
dass es 4 aktive Orbitale gibt. 11 und 12 sind offene Orbitale. Sie bilden ein
resonantes ungepaartes Elektronenpaar (d. h. NSETO=2 bedeutet ein radikalisches
Elektronenpaar (2 radikalische Elektronen). 13 und 14 bilden ein resonantes
spin-gepaartes Elektronenpaar.
----------------------------
ROHF-GVB COUPLING PARAMETERS
----------------------------
F VECTOR (OCCUPANCIES)
1 1.0000000000
2 0.5000000000
3 0.5000000000
4 0.9529411765
5 0.0470588235
ALPHA COUPLING COEFFICEINTS
1 2 3 4 5
1 2.0000000
2 1.0000000 0.0000000
3 1.0000000 0.5000000 0.0000000
4 1.9058824 0.9529412 0.9529412 0.9529412
5 0.0941176 0.0470588 0.0470588 0.0000000 0.0470588
BETA COUPLING COEFFICIENTS
1 2 3 4 5
1 -1.0000000
2 -0.5000000 0.0000000
3 -0.5000000 0.5000000 0.0000000
4 -0.9529412 -0.4764706 -0.4764706 0.0000000
5 -0.0470588 -0.0235294 -0.0235294 -0.2117647 0.0000000
NATURAL ORBITAL COEFFICIENTS
N.O. PAIR CICOEF-S
1 0.9761870602 -0.2169304578
Mit
$SCF NCO=10 NPAIR=2 $END
$GUESS GUESS=MOREAD NORB=15 $END
Ab NPAIR=2 ist GUESS=MOREAD notwendig! Ebenfalls hier MAXIT=50 in $CONTRL!
*************************
ROHF-GVB INPUT PARAMETERS
*************************
NORB = 14 NCO = 10
NPAIR = 2 NSETO = 0
PAIR ORBITALS
PAIR 1 HAS ORBS 11 12
PAIR 2 HAS ORBS 13 14
2 spin-gepaarte Elektronenpaare
----------------------------
ROHF-GVB COUPLING PARAMETERS
----------------------------
F VECTOR (OCCUPANCIES)
1 1.0000000000
2 0.9848769847
3 0.0151230153
4 0.9529411765
5 0.0470588235
ALPHA COUPLING COEFFICEINTS
1 2 3 4 5
1 2.0000000
2 1.9697540 0.9848770
3 0.0302460 0.0000000 0.0151230
4 1.9058824 1.8770597 0.0288227 0.9529412
5 0.0941176 0.0926943 0.0014233 0.0000000 0.0470588
BETA COUPLING COEFFICIENTS
1 2 3 4 5
1 -1.0000000
2 -0.9848770 0.0000000
3 -0.0151230 -0.1220422 0.0000000
4 -0.9529412 -0.9385298 -0.0144113 0.0000000
5 -0.0470588 -0.0463472 -0.0007117 -0.2117647 0.0000000
NATURAL ORBITAL COEFFICIENTS
N.O. PAIR CICOEF-S
1 0.9924096859 -0.1229756695
2 0.9761870602 -0.2169304578
------------------------
ROHF-GVB SCF CALCULATION
------------------------
GVB STEP WILL USE 52664 WORDS OF MEMORY.
MAXIT= 30 NPUNCH= 2 SQCDF TOL=1.0000E-05
NUCLEAR ENERGY= 68.5566738453
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=T
SOSCF WILL OPTIMIZE 36 ORBITAL ROTATIONS, SOGTOL= 0.250
ITER EX TOTAL ENERGY E CHANGE SQCDF ORB. GRAD
0 0 -222.803005860 -222.803005860 0.540263691 0.000000000
---------------START SECOND ORDER SCF---------------
1 1 -223.320164040 -0.517158181 0.070211892 0.160622584
2 2 -223.391543602 -0.071379561 0.008857215 0.081085983
3 3 -223.415655843 -0.024112241 0.000182287 0.010706273
4 4 -223.416212375 -0.000556532 0.000077314 0.003576370
5 5 -223.416322926 -0.000110551 0.000004301 0.000988809
6 6 -223.416330438 -0.000007513 0.000000602 0.000345970
7 7 -223.416331362 -0.000000924 0.000000022 0.000098404
8 8 -223.416331416 -0.000000054 0.000000012 0.000059246
9 9 -223.416331432 -0.000000017 0.000000001 0.000015910
10 10 -223.416331434 -0.000000002 0.000000000 0.000007061
11 11 -223.416331435 0.000000000 0.000000000 0.000002134
12 12 -223.416331435 0.000000000 0.000000000 0.000001008
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -223.4163314346 AFTER 12 ITERATIONS
GVB STEP WILL USE 54045 WORDS OF MEMORY.
MAXIT= 30 NPUNCH= 2 SQCDF TOL=1.0000E-05
NUCLEAR ENERGY= 68.5566738453
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=T
SOSCF WILL OPTIMIZE 49 ORBITAL ROTATIONS, SOGTOL= 0.250
ITER EX TOTAL ENERGY E CHANGE SQCDF ORB. GRAD
0 0 -222.807572637 -222.807572637 0.606365047 0.480459502
---------------START SECOND ORDER SCF---------------
1 1 -223.338291423 -0.530718786 0.179292357 0.152513146
2 2 -223.375222781 -0.036931358 0.033572090 0.111620109
3 3 -223.418271132 -0.043048351 0.000622725 0.014922891
4 4 -223.418941241 -0.000670109 0.000163280 0.004742637
5 5 -223.419015559 -0.000074318 0.000002237 0.000752066
6 6 -223.419019327 -0.000003768 0.000003868 0.000306412
7 7 -223.419020280 -0.000000952 0.000001335 0.000069565
8 8 -223.419020320 -0.000000041 0.000000911 0.000041209
9 9 -223.419020332 -0.000000012 0.000000028 0.000011185
10 10 -223.419020333 -0.000000001 0.000000001 0.000004725
11 11 -223.419020334 0.000000000 0.000000000 0.000001210
-----------------
DENSITY CONVERGED
-----------------
GVB STEP WILL USE 54045 WORDS OF MEMORY.
MAXIT= 30 NPUNCH= 2 SQCDF TOL=1.0000E-05
NUCLEAR ENERGY= 68.5566738453
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=T
SOSCF WILL OPTIMIZE 49 ORBITAL ROTATIONS, SOGTOL= 0.250
ITER EX TOTAL ENERGY E CHANGE SQCDF ORB. GRAD
0 0 -222.754142450 -222.754142450 0.617305403 0.000000000
---------------START SECOND ORDER SCF---------------
1 1 -223.315386005 -0.561243555 0.194698708 0.128461614
2 2 -223.339269628 -0.023883623 0.056276167 0.066876257
3 3 -223.372274464 -0.033004836 0.004063123 0.006896342
4 4 -223.373870264 -0.001595800 0.005291585 0.003962423
5 5 -223.374492693 -0.000622429 0.000108734 0.003293219
6 6 -223.374556355 -0.000063662 0.000231484 0.001634742
7 7 -223.374591817 -0.000035463 0.000003513 0.000359111
8 8 -223.374593369 -0.000001551 0.000001339 0.000184459
9 9 -223.374593805 -0.000000436 0.000000003 0.000036125
10 10 -223.374593812 -0.000000006 0.000000001 0.000010000
11 11 -223.374593812 -0.000000001 0.000000000 0.000002877
12 12 -223.374593812 0.000000000 0.000000000 0.000001301
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -223.3745938123 AFTER 12 ITERATIONS
GVB STEP WILL USE 55412 WORDS OF MEMORY.
MAXIT= 30 NPUNCH= 2 SQCDF TOL=1.0000E-05
NUCLEAR ENERGY= 68.5566738453
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=T
SOSCF WILL OPTIMIZE 60 ORBITAL ROTATIONS, SOGTOL= 0.250
ITER EX TOTAL ENERGY E CHANGE SQCDF ORB. GRAD
0 0 -222.658144419 -222.658144419 0.702906076 0.484198509
---------------START SECOND ORDER SCF---------------
1 1 -223.190041089 -0.531896670 0.146858492 0.083742371
2 2 -223.205209169 -0.015168081 0.038018994 0.055014833
3 3 -223.234927568 -0.029718399 0.001064142 0.010133059
4 4 -223.235536531 -0.000608963 0.000888633 0.003136165
5 5 -223.235687950 -0.000151419 0.000548635 0.001083699
6 6 -223.235724580 -0.000036629 0.000923797 0.000966190
7 7 -223.235750644 -0.000026065 0.001005611 0.001175557
8 8 -223.235768430 -0.000017786 0.000327567 0.000820178
9 9 -223.235775394 -0.000006964 0.000007577 0.000358266
10 10 -223.235776365 -0.000000971 0.000000357 0.000063524
11 11 -223.235776415 -0.000000050 0.000000396 0.000009024
12 12 -223.235776422 -0.000000007 0.000004262 0.000007483
13 13 -223.235776433 -0.000000011 0.000009373 0.000009782
14 14 -223.235776441 -0.000000009 0.000001798 0.000009075
15 15 -223.235776443 -0.000000002 0.000000016 0.000003212
16 16 -223.235776443 0.000000000 0.000000002 0.000000396
17 17 -223.235776443 0.000000000 0.000000000 0.000000125
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -223.2357764435 AFTER 17 ITERATIONS
GVB STEP WILL USE 55412 WORDS OF MEMORY.
MAXIT= 50 NPUNCH= 2 SQCDF TOL=1.0000E-05
NUCLEAR ENERGY= 68.5566738453
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=T
SOSCF WILL OPTIMIZE 60 ORBITAL ROTATIONS, SOGTOL= 0.250
ITER EX TOTAL ENERGY E CHANGE SQCDF ORB. GRAD
0 0 -223.347719150 -223.347719150 0.789062767 0.000000000
---------------START SECOND ORDER SCF---------------
SOSCF IS SCALING ROTATION ANGLE MATRIX, SQCDF= 0.100728
1 1 -220.989167117 2.358552033 0.925042419 0.125227373
2 2 -226.859946323 -5.870779206 0.237646890 0.144625135
3 3 -225.324409286 1.535537037 0.132819851 0.034648987
4 4 -225.531527738 -0.207118452 0.030858488 0.015750662
5 5 -225.373430488 0.158097251 0.065752757 0.010514439
6 6 -224.720281864 0.653148624 0.787101121 0.015162531
7 7 -223.361494078 1.358787786 0.233545358 0.077663598
8 8 -224.192620443 -0.831126365 0.091592989 0.030321938
9 9 -224.131859582 0.060760862 0.571404308 0.020645032
10 10 -223.357503481 0.774356101 0.273813401 0.044831320
11 11 -223.741062582 -0.383559102 0.045222583 0.038908178
12 12 -223.655988668 0.085073915 0.012484678 0.027299023
13 13 -223.598599234 0.057389433 0.002071754 0.011740721
14 14 -223.616338516 -0.017739282 0.010148934 0.002007255
15 15 -223.601354395 0.014984122 0.000852544 0.005792249
16 16 -223.637880270 -0.036525875 0.001080540 0.003538966
17 17 -223.645165866 -0.007285596 0.000122307 0.002706588
18 18 -223.647802706 -0.002636840 0.000416524 0.000542787
19 19 -223.645636625 0.002166081 0.000028030 0.001059387
20 20 -223.646630771 -0.000994146 0.000115717 0.000306736
21 21 -223.647473931 -0.000843160 0.000356427 0.000037837
22 22 -223.646407107 0.001066824 0.000174437 0.000099420
23 23 -223.645773977 0.000633130 0.000019575 0.000093781
24 24 -223.644068166 0.001705812 0.000000583 0.000026089
25 25 -223.644241828 -0.000173663 0.000000761 0.000015012
26 26 -223.644080997 0.000160831 0.000000216 0.000013553
27 27 -223.644126711 -0.000045713 0.000000299 0.000007845
28 28 -223.644282101 -0.000155391 0.000000041 0.000005632
29 29 -223.644332218 -0.000050117 0.000000550 0.000005878
30 30 -223.644381475 -0.000049257 0.000000160 0.000006234
31 31 -223.644337995 0.000043480 0.000000352 0.000003189
32 32 -223.644262552 0.000075443 0.000000004 0.000002424
33 33 -223.644268828 -0.000006277 0.000000007 0.000001812
34 34 -223.644296880 -0.000028051 0.000000001 0.000000661
35 35 -223.644304006 -0.000007126 0.000000005 0.000000497
36 36 -223.644309144 -0.000005138 0.000000002 0.000000197
37 37 -223.644306611 0.000002534 0.000000000 0.000000127
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -223.6443066107 AFTER 37 ITERATIONS
FINAL ENERGY IS -223.4163314346 AFTER 12 ITERATIONS
FINAL ENERGY IS -223.4190203335 AFTER 11 ITERATIONS
FINAL ENERGY IS -223.3745938123
AFTER 12 ITERATIONS
FINAL ENERGY IS -223.2357764435
AFTER 17 ITERATIONS
FINAL ENERGY IS -223.6443066107
AFTER 37 ITERATIONS
Die Einbeziehung von spin-gepaarten
Grenzstrukturen führt zu einer Erniedrigung der Gesamtenergie (NPAIR=1,
NPAIR=2).
Radikalische Grenzstrukturen führen dagegen offensichtlich zur Destabilisierung.
Zum weiteren Vergleich hier die RHF-Rechnung
(immer in
dunkelblau)
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 68.5566738453
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=T
DENSITY CONV= 1.00E-05
SOSCF WILL OPTIMIZE 36 ORBITAL ROTATIONS, SOGTOL= 0.250
MEMORY REQUIRED FOR RHF STEP= 9426 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE ORB. GRAD
1 0 0 -222.803005860 -222.803005860 0.648354295 0.000000000
---------------START SECOND ORDER SCF---------------
2 1 0 -223.320164040 -0.517158181 0.302627540 0.160622584
3 2 0 -223.402840917 -0.082676877 0.086926518 0.058904514
4 3 0 -223.415286665 -0.012445748 0.029076858 0.010015109
5 4 0 -223.416111993 -0.000825328 0.018578230 0.005152052
6 5 0 -223.416308085 -0.000196092 0.004168454 0.001355881
7 6 0 -223.416323365 -0.000015281 0.004010740 0.001000308
8 7 0 -223.416331016 -0.000007651 0.000843500 0.000211913
9 8 0 -223.416331395 -0.000000378 0.000234284 0.000082593
10 9 0 -223.416331434 -0.000000039 0.000025162 0.000015370
11 10 0 -223.416331435 -0.000000001 0.000006307 0.000001968
12 11 0 -223.416331435 0.000000000 0.000001573 0.000000542
-----------------
DENSITY CONVERGED
-----------------
TIME TO FORM FOCK OPERATORS= 0.1 SECONDS ( 0.0 SEC/ITER)
TIME TO SOLVE SCF EQUATIONS= 0.0 SECONDS ( 0.0 SEC/ITER)
FINAL ENERGY IS -223.4163314346 AFTER 12 ITERATIONS
Vergleich mit GVB von oben:
FINAL ENERGY IS -223.4163314346 AFTER 12 ITERATIONS
FINAL ENERGY IS -223.4190203335 AFTER 11 ITERATIONS
MO
und VB mit "normalen" Resonanzstrukturen ergeben das gleiche Resultat!
----------------------------
SCF STATISTICS PER ITERATION
----------------------------
NUMBER OF INTEGRAL PASSES 1
FOCK FORMATION TIME 0.004
GEMINAL OPT TIME 0.000
MIXORB OPT TIME 0.000
OCBSE OPT TIME 0.000
THE MAXIMUM LAGRANGIAN ASYMMETRY IS 1.7112293E-16
NUMBER OF INTEGRAL PASSES 1
FOCK FORMATION TIME 0.005
GEMINAL OPT TIME 0.000
MIXORB OPT TIME 0.000
OCBSE OPT TIME 0.000
----------------------------
SCF STATISTICS PER ITERATION
----------------------------
NUMBER OF INTEGRAL PASSES 1
FOCK FORMATION TIME 0.009
GEMINAL OPT TIME 0.000
MIXORB OPT TIME 0.000
OCBSE OPT TIME 0.000
THE MAXIMUM LAGRANGIAN ASYMMETRY IS 3.0996686E-07
----------------------------
SCF STATISTICS PER ITERATION
----------------------------
NUMBER OF INTEGRAL PASSES 1
FOCK FORMATION TIME 0.006
GEMINAL OPT TIME 0.000
MIXORB OPT TIME 0.000
OCBSE OPT TIME 0.000
----------------
PAIR INFORMATION
----------------
ORBITAL CI COEFFICIENTS OCCUPATION NUMBERS GVB ENERGY
PAIR 1 2 ORB 1 ORB 2 ORB 1 ORB 2 OVERLAP LOWERING
1 13 14 0.999722 -0.023586 1.99889 0.00111 0.95390 -0.00057
THE MAXIMUM LAGRANGIAN ASYMMETRY IS 1.3310022E-07
ORBITAL CI COEFFICIENTS OCCUPATION NUMBERS GVB ENERGY
PAIR 1 2 ORB 1 ORB 2 ORB 1 ORB 2 OVERLAP LOWERING
1 12 13 0.992410 -0.122976 1.96975 0.03025 0.77949 -0.00269
THE MAXIMUM LAGRANGIAN ASYMMETRY IS 9.8187071E-06
------------
EIGENVECTORS
------------
1 2 3 4 5
-20.9018 -20.6184 -20.6183 -1.6502 -1.3192
A1 B2 A1 A1 B2
1 O 1 S 0.996922 0.000000 0.000426 -0.185325 0.000000
2 O 1 S 0.013916 0.000000 -0.003692 0.684988 0.000000
3 O 1 X 0.000000 0.000000 0.000000 0.000000 0.000000
4 O 1 Y 0.000000 0.002698 0.000000 0.000000 -0.315234
5 O 1 Z 0.002454 0.000000 -0.001611 0.132081 0.000000
6 O 2 S 0.000244 0.704968 0.704995 -0.092708 -0.156168
7 O 2 S -0.002366 0.009381 0.009292 0.315110 0.608905
8 O 2 X 0.000000 0.000000 0.000000 0.000000 0.000000
9 O 2 Y -0.001731 0.001588 0.001520 0.091906 0.067726
10 O 2 Z 0.000893 -0.000927 -0.000930 -0.048178 -0.058627
11 O 3 S 0.000244 -0.704968 0.704995 -0.092708 0.156168
12 O 3 S -0.002366 -0.009381 0.009292 0.315110 -0.608905
13 O 3 X 0.000000 0.000000 0.000000 0.000000 0.000000
14 O 3 Y 0.001731 0.001588 -0.001520 -0.091906 0.067726
15 O 3 Z 0.000893 0.000927 -0.000930 -0.048178 0.058627
6 7 8 9 10
-0.9892 -0.6963 -0.6637 -0.6570 -0.4263
A1 A1 B1 B2 B2
1 O 1 S -0.150260 0.015333 0.000000 0.000000 0.000000
2 O 1 S 0.690537 -0.081227 0.000000 0.000000 0.000000
3 O 1 X 0.000000 0.000000 0.762281 0.000000 0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.581612 -0.145482
5 O 1 Z -0.226236 0.692988 0.000000 0.000000 0.000000
6 O 2 S 0.138406 0.054151 0.000000 -0.079225 0.010752
7 O 2 S -0.609707 -0.269367 0.000000 0.397353 -0.059809
8 O 2 X 0.000000 0.000000 0.370011 0.000000 0.000000
9 O 2 Y 0.067530 0.347492 0.000000 -0.222489 0.507732
10 O 2 Z -0.098334 0.163224 0.000000 0.347664 0.497122
11 O 3 S 0.138406 0.054151 0.000000 0.079225 -0.010752
12 O 3 S -0.609707 -0.269367 0.000000 -0.397353 0.059809
13 O 3 X 0.000000 0.000000 0.370011 0.000000 0.000000
14 O 3 Y -0.067530 -0.347492 0.000000 -0.222489 0.507732
15 O 3 Z -0.098334 0.163224 0.000000 -0.347664 -0.497122
11 12 13 14 15
-0.4033 -0.3320 0.0931 0.5040 0.6140
A1 A2 B1 A1 B2
1 O 1 S -0.035471 0.000000 0.000000 -0.114159 0.000000
2 O 1 S 0.200155 0.000000 0.000000 0.716595 0.000000
3 O 1 X 0.000000 0.000000 0.672009 0.000000 0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.000000 0.953715
5 O 1 Z -0.447005 0.000000 0.000000 0.641900 0.000000
6 O 2 S -0.007562 0.000000 0.000000 0.050559 -0.052883
7 O 2 S 0.047423 0.000000 0.000000 -0.305407 0.331653
8 O 2 X 0.000000 0.710430 -0.612089 0.000000 0.000000
9 O 2 Y 0.142003 0.000000 0.000000 -0.667183 0.521725
10 O 2 Z 0.639516 0.000000 0.000000 0.280850 -0.426832
11 O 3 S -0.007562 0.000000 0.000000 0.050559 0.052883
12 O 3 S 0.047423 0.000000 0.000000 -0.305407 -0.331653
13 O 3 X 0.000000 -0.710430 -0.612089 0.000000 0.000000
14 O 3 Y -0.142003 0.000000 0.000000 0.667183 0.521725
15 O 3 Z 0.639516 0.000000 0.000000 0.280850 0.426832
... END OF ROHF-GVB SCF CALCULATION ...
1 2 3 4 5
-20.6179 -20.6179 -20.9018 -1.6471 -1.3189
A1 B2 A1 A1 B2
1 O 1 S 0.000429 0.000000 0.996989 -0.181831 0.000000
2 O 1 S -0.003692 0.000000 0.013572 0.667336 0.000000
3 O 1 X 0.000000 0.000000 0.000000 0.000000 0.000000
4 O 1 Y 0.000000 -0.002699 0.000000 0.000000 -0.315478
5 O 1 Z -0.001612 0.000000 0.003012 0.159222 0.000000
6 O 2 S 0.704995 -0.704968 0.000243 -0.093326 -0.156151
7 O 2 S 0.009292 -0.009381 -0.002385 0.317101 0.608806
8 O 2 X 0.000000 0.000000 0.000000 0.000000 0.000000
9 O 2 Y 0.001520 -0.001587 -0.001850 0.086883 0.067721
10 O 2 Z -0.000930 0.000927 0.000261 -0.075540 -0.058486
11 O 3 S 0.704995 0.704968 0.000243 -0.093326 0.156151
12 O 3 S 0.009292 0.009381 -0.002385 0.317101 -0.608806
13 O 3 X 0.000000 0.000000 0.000000 0.000000 0.000000
14 O 3 Y -0.001520 -0.001587 0.001850 -0.086883 0.067721
15 O 3 Z -0.000930 -0.000927 0.000261 -0.075540 0.058486
6 7 8 9 10
-0.9807 -0.6646 -0.6917 -0.6565 -0.3338
A1 B1 A1 B2 A2
1 O 1 S -0.145543 0.000000 0.013665 0.000000 0.000000
2 O 1 S 0.663514 0.000000 -0.069155 0.000000 0.000000
3 O 1 X 0.000000 0.761821 0.000000 0.000000 0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.582857 0.000000
5 O 1 Z -0.153304 0.000000 0.640693 0.000000 0.000000
6 O 2 S 0.139348 0.000000 0.051095 -0.079315 0.000000
7 O 2 S -0.616771 0.000000 -0.253937 0.397863 0.000000
8 O 2 X 0.000000 0.370431 0.000000 0.000000 0.710430
9 O 2 Y 0.060416 0.000000 0.360557 -0.223965 0.000000
10 O 2 Z -0.167428 0.000000 0.235635 0.345489 0.000000
11 O 3 S 0.139348 0.000000 0.051095 0.079315 0.000000
12 O 3 S -0.616771 0.000000 -0.253937 -0.397863 0.000000
13 O 3 X 0.000000 0.370431 0.000000 0.000000 -0.710430
14 O 3 Y -0.060416 0.000000 -0.360557 -0.223965 0.000000
15 O 3 Z -0.167428 0.000000 0.235635 -0.345489 0.000000
11 12 13 14 15
-0.4247 -0.4078 -0.0001 0.5046 0.6143
B2 A1 B1 A1 B2
1 O 1 S 0.000000 -0.062163 0.000000 -0.114101 0.000000
2 O 1 S 0.000000 0.320563 0.000000 0.716307 0.000000
3 O 1 X 0.000000 0.000000 0.672532 0.000000 0.000000
4 O 1 Y -0.142245 0.000000 0.000000 0.000000 0.953362
5 O 1 Z 0.000000 -0.536948 0.000000 0.642691 0.000000
6 O 2 S 0.010427 -0.001478 0.000000 0.050562 -0.052863
7 O 2 S -0.058078 0.019338 0.000000 -0.305453 0.331529
8 O 2 X 0.000000 0.000000 -0.611835 0.000000 0.000000
9 O 2 Y 0.507457 0.113477 0.000000 -0.667067 0.521362
10 O 2 Z 0.497954 0.598767 0.000000 0.280547 -0.427646
11 O 3 S -0.010427 -0.001478 0.000000 0.050562 0.052863
12 O 3 S 0.058078 0.019338 0.000000 -0.305453 -0.331529
13 O 3 X 0.000000 0.000000 -0.611835 0.000000 0.000000
14 O 3 Y 0.507457 -0.113477 0.000000 0.667067 0.521362
15 O 3 Z -0.497954 0.598767 0.000000 0.280547 0.427646
-----------
GI ORBITALS
-----------
PAIR 1
1 2
1 O 1 S -0.058636 0.058636
2 O 1 S 0.302375 -0.302375
3 O 1 X 0.223311 0.223311
4 O 1 Y 0.000000 0.000000
5 O 1 Z -0.506484 0.506484
6 O 2 S -0.001394 0.001394
7 O 2 S 0.018240 -0.018240
8 O 2 X -0.203157 -0.203157
9 O 2 Y 0.107039 -0.107039
10 O 2 Z 0.564795 -0.564795
11 O 3 S -0.001394 0.001394
12 O 3 S 0.018240 -0.018240
13 O 3 X -0.203157 -0.203157
14 O 3 Y -0.107039 0.107039
15 O 3 Z 0.564795 -0.564795
... END OF ROHF-GVB SCF CALCULATION ...
------------
EIGENVECTORS
------------
1 2 3 4 5
-20.6068 -20.6068 -20.8422 -1.6183 -1.3015
A1 B2 A1 A1 B2
1 O 1 S 0.000946 0.000000 0.996882 -0.183707 0.000000
2 O 1 S -0.003680 0.000000 0.014084 0.676552 0.000000
3 O 1 X 0.000000 0.000000 0.000000 0.000000 0.000000
4 O 1 Y 0.000000 0.002694 0.000000 0.000000 -0.299580
5 O 1 Z -0.001682 0.000000 0.002156 0.114788 0.000000
6 O 2 S 0.704985 0.704966 -0.000112 -0.095275 -0.157765
7 O 2 S 0.009334 0.009391 -0.002397 0.327730 0.617375
8 O 2 X 0.000000 0.000000 0.000000 0.000000 0.000000
9 O 2 Y 0.001426 0.001458 -0.001741 0.089966 0.059910
10 O 2 Z -0.000885 -0.000922 0.000871 -0.048533 -0.061803
11 O 3 S 0.704985 -0.704966 -0.000112 -0.095275 0.157765
12 O 3 S 0.009334 -0.009391 -0.002397 0.327730 -0.617375
13 O 3 X 0.000000 0.000000 0.000000 0.000000 0.000000
14 O 3 Y -0.001426 0.001458 0.001741 -0.089966 0.059910
15 O 3 Z -0.000885 0.000922 0.000871 -0.048533 0.061803
6 7 8 9 10
-0.9670 -0.6771 -0.5287 -0.6209 -0.4581
A1 B1 A1 B2 A2
1 O 1 S -0.148578 0.000000 0.000726 0.000000 0.000000
2 O 1 S 0.678409 0.000000 0.007303 0.000000 0.000000
3 O 1 X 0.000000 0.540367 0.000000 0.000000 0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.573654 0.000000
5 O 1 Z -0.116140 0.000000 0.428916 0.000000 0.000000
6 O 2 S 0.142835 0.000000 0.036218 -0.075788 0.000000
7 O 2 S -0.634938 0.000000 -0.182772 0.382167 0.000000
8 O 2 X 0.000000 0.528428 0.000000 0.000000 0.710430
9 O 2 Y 0.086257 0.000000 0.356089 -0.162125 0.000000
10 O 2 Z -0.123084 0.000000 0.454625 0.397903 0.000000
11 O 3 S 0.142835 0.000000 0.036218 0.075788 0.000000
12 O 3 S -0.634938 0.000000 -0.182772 -0.382167 0.000000
13 O 3 X 0.000000 0.528428 0.000000 0.000000 -0.710430
14 O 3 Y -0.086257 0.000000 -0.356089 -0.162125 0.000000
15 O 3 Z -0.123084 0.000000 0.454625 -0.397903 0.000000
11 12 13 14 15
-0.2120 -0.2279 -0.3709 0.0000 0.5398
B2 A1 B1 B2 A1
1 O 1 S 0.000000 0.064122 0.000000 0.000000 -0.107665
2 O 1 S 0.000000 -0.353248 0.000000 0.000000 0.680745
3 O 1 X 0.000000 0.000000 0.860624 0.000000 0.000000
4 O 1 Y -0.108491 0.000000 0.000000 0.968393 0.000000
5 O 1 Z 0.000000 0.668771 0.000000 0.000000 0.709100
6 O 2 S 0.011449 0.010543 0.000000 -0.053067 0.049069
7 O 2 S -0.057992 -0.065031 0.000000 0.334152 -0.301545
8 O 2 X 0.000000 0.000000 -0.482001 0.000000 0.000000
9 O 2 Y 0.583862 0.099938 0.000000 0.461853 -0.668382
10 O 2 Z 0.405418 -0.503594 0.000000 -0.476563 0.220667
11 O 3 S -0.011449 0.010543 0.000000 0.053067 0.049069
12 O 3 S 0.057992 -0.065031 0.000000 -0.334152 -0.301545
13 O 3 X 0.000000 0.000000 -0.482001 0.000000 0.000000
14 O 3 Y 0.583862 -0.099938 0.000000 0.461853 0.668382
15 O 3 Z -0.405418 -0.503594 0.000000 0.476563 0.220667
-----------
GI ORBITALS
-----------
PAIR 1
1 2
1 O 1 S 0.000000 0.000000
2 O 1 S 0.000000 0.000000
3 O 1 X 0.850648 -0.850648
4 O 1 Y 0.147021 0.147021
5 O 1 Z 0.000000 0.000000
6 O 2 S -0.008057 -0.008057
7 O 2 S 0.050731 0.050731
8 O 2 X -0.476414 0.476414
9 O 2 Y 0.070118 0.070118
10 O 2 Z -0.072351 -0.072351
11 O 3 S 0.008057 0.008057
12 O 3 S -0.050731 -0.050731
13 O 3 X -0.476414 0.476414
14 O 3 Y 0.070118 0.070118
15 O 3 Z 0.072351 0.072351
... END OF ROHF-GVB SCF CALCULATION ...
------------
EIGENVECTORS
------------
1 2 3 4 5
-20.9018 -20.6184 -20.6183 -1.6502 -1.3192
A1 B2 A1 A1 B2
1 O 1 S 0.996922 0.000000 0.000426 -0.185325 0.000000
2 O 1 S 0.013916 0.000000 -0.003692 0.684988 0.000000
3 O 1 X 0.000000 0.000000 0.000000 0.000000 0.000000
4 O 1 Y 0.000000 0.002698 0.000000 0.000000 -0.315234
5 O 1 Z 0.002454 0.000000 -0.001611 0.132081 0.000000
6 O 2 S 0.000244 0.704968 0.704995 -0.092708 -0.156168
7 O 2 S -0.002366 0.009381 0.009292 0.315110 0.608905
8 O 2 X 0.000000 0.000000 0.000000 0.000000 0.000000
9 O 2 Y -0.001731 0.001588 0.001520 0.091906 0.067726
10 O 2 Z 0.000893 -0.000927 -0.000930 -0.048178 -0.058627
11 O 3 S 0.000244 -0.704968 0.704995 -0.092708 0.156168
12 O 3 S -0.002366 -0.009381 0.009292 0.315110 -0.608905
13 O 3 X 0.000000 0.000000 0.000000 0.000000 0.000000
14 O 3 Y 0.001731 0.001588 -0.001520 -0.091906 0.067726
15 O 3 Z 0.000893 0.000927 -0.000930 -0.048178 0.058627
6 7 8 9 10
-0.9892 -0.6963 -0.6637 -0.6570 -0.4263
A1 A1 B1 B2 B2
1 O 1 S -0.150260 0.015333 0.000000 0.000000 0.000000
2 O 1 S 0.690537 -0.081227 0.000000 0.000000 0.000000
3 O 1 X 0.000000 0.000000 0.762281 0.000000 0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.581612 -0.145481
5 O 1 Z -0.226236 0.692987 0.000000 0.000000 0.000000
6 O 2 S 0.138406 0.054151 0.000000 -0.079225 0.010752
7 O 2 S -0.609707 -0.269367 0.000000 0.397353 -0.059809
8 O 2 X 0.000000 0.000000 0.370011 0.000000 0.000000
9 O 2 Y 0.067530 0.347492 0.000000 -0.222489 0.507732
10 O 2 Z -0.098334 0.163224 0.000000 0.347663 0.497122
11 O 3 S 0.138406 0.054151 0.000000 0.079225 -0.010752
12 O 3 S -0.609707 -0.269367 0.000000 -0.397353 0.059809
13 O 3 X 0.000000 0.000000 0.370011 0.000000 0.000000
14 O 3 Y -0.067530 -0.347492 0.000000 -0.222489 0.507732
15 O 3 Z -0.098334 0.163224 0.000000 -0.347663 -0.497122
11 12 13 14 15
-0.4033 -0.3320 0.0931 0.5040 0.6140
A1 A2 B1 A1 B2
1 O 1 S -0.035471 0.000000 0.000000 -0.114159 0.000000
2 O 1 S 0.200155 0.000000 0.000000 0.716595 0.000000
3 O 1 X 0.000000 0.000000 0.672009 0.000000 0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.000000 0.953715
5 O 1 Z -0.447006 0.000000 0.000000 0.641900 0.000000
6 O 2 S -0.007562 0.000000 0.000000 0.050559 -0.052883
7 O 2 S 0.047423 0.000000 0.000000 -0.305408 0.331653
8 O 2 X 0.000000 0.710430 -0.612089 0.000000 0.000000
9 O 2 Y 0.142003 0.000000 0.000000 -0.667183 0.521725
10 O 2 Z 0.639515 0.000000 0.000000 0.280850 -0.426832
11 O 3 S -0.007562 0.000000 0.000000 0.050559 0.052883
12 O 3 S 0.047423 0.000000 0.000000 -0.305408 -0.331653
13 O 3 X 0.000000 -0.710430 -0.612089 0.000000 0.000000
14 O 3 Y -0.142003 0.000000 0.000000 0.667183 0.521725
15 O 3 Z 0.639515 0.000000 0.000000 0.280850 0.426832
...... END OF RHF CALCULATION ......
------------------------------
properties for the GVB density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -445.3942187160
TWO ELECTRON ENERGY = 153.4212134361
NUCLEAR REPULSION ENERGY = 68.5566738453
------------------
TOTAL ENERGY = -223.4163314346
ELECTRON-ELECTRON POTENTIAL ENERGY = 153.4212134361
NUCLEUS-ELECTRON POTENTIAL ENERGY = -668.2974391096
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 68.5566738453
------------------
TOTAL POTENTIAL ENERGY = -446.3195518282
TOTAL KINETIC ENERGY = 222.9032203936
VIRIAL RATIO (V/T) = 2.0023019454
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -445.3967863180
TWO ELECTRON ENERGY = 153.4210921392
NUCLEAR REPULSION ENERGY = 68.5566738453
------------------
TOTAL ENERGY = -223.4190203335
ELECTRON-ELECTRON POTENTIAL ENERGY = 153.4210921392
NUCLEUS-ELECTRON POTENTIAL ENERGY = -668.3042058178
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 68.5566738453
------------------
TOTAL POTENTIAL ENERGY = -446.3264398333
TOTAL KINETIC ENERGY = 222.9074194998
VIRIAL RATIO (V/T) = 2.0022951270
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -445.5069768778
TWO ELECTRON ENERGY = 153.7145265890
NUCLEAR REPULSION ENERGY = 68.5566738453
------------------
TOTAL ENERGY = -223.2357764435
ELECTRON-ELECTRON POTENTIAL ENERGY = 153.7145265890
NUCLEUS-ELECTRON POTENTIAL ENERGY = -668.9755574238
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 68.5566738453
------------------
TOTAL POTENTIAL ENERGY = -446.7043569895
TOTAL KINETIC ENERGY = 223.4685805460
VIRIAL RATIO (V/T) = 1.9989582245
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -445.3942187251
TWO ELECTRON ENERGY = 153.4212134452
NUCLEAR REPULSION ENERGY = 68.5566738453
------------------
TOTAL ENERGY = -223.4163314346
ELECTRON-ELECTRON POTENTIAL ENERGY = 153.4212134452
NUCLEUS-ELECTRON POTENTIAL ENERGY = -668.2974387516
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 68.5566738453
------------------
TOTAL POTENTIAL ENERGY = -446.3195514611
TOTAL KINETIC ENERGY = 222.9032200265
VIRIAL RATIO (V/T) = 2.0023019470
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -138.5517910247
BARE H ENERGY= -445.3942187251
ELECTRONIC ENERGY = -291.9730048749
KINETIC ENERGY= 222.9032200265
N-N REPULSION= 68.5566738453
TOTAL ENERGY= -223.4163310296
SIGMA PART(1+2)= -267.1116627633
(K,V1,2)= 213.3678621275 -611.0308970179 130.5513721271
PI PART(1+2)= -24.8613421116
(K,V1,2)= 9.5353578990 -57.2665417337 22.8698417231
SIGMA SKELETON, ERROR= -198.5549889180 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 2.000447 -0.000219 -0.000286 1.240898 0.408866
2 -0.000223 1.000109 1.000143 0.379551 0.795567
3 -0.000223 1.000109 1.000143 0.379551 0.795567
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.755337 1.051386 1.304701 0.700791 0.032481
2 0.622332 0.474307 0.347650 0.649604 0.983760
3 0.622332 0.474307 0.347650 0.649604 0.983760
11 12
2.000000 2.000000
1 0.364263 0.000000
2 0.817868 1.000000
3 0.817868 1.000000
MULLIKEN ATOMIC POPULATION
IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 -0.000286 -0.000219 2.000417 1.219058 0.409272
2 1.000143 1.000109 -0.000209 0.390471 0.795364
3 1.000143 1.000109 -0.000209 0.390471 0.795364
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.640329 1.303372 0.917542 0.703122 0.000000
2 0.679835 0.348314 0.541229 0.648439 1.000000
3 0.679835 0.348314 0.541229 0.648439 1.000000
11 12 13
2.000000 1.969754 0.030246
1 0.030955 0.624159 0.010535
2 0.984522 0.672797 0.009855
3 0.984522 0.672797 0.009855
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 -0.000289 -0.000218 2.000448 1.210580 0.380640
2 1.000144 1.000109 -0.000224 0.394710 0.809680
3 1.000144 1.000109 -0.000224 0.394710 0.809680
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.625691 0.728312 0.444586 0.687244 0.000000
2 0.687155 0.635844 0.777707 0.656378 1.000000
3 0.687155 0.635844 0.777707 0.656378 1.000000
11 12 13 14
1.000000 1.000000 1.998887 0.001113
1 0.014866 0.512710 1.270980 0.000502
2 0.492567 0.243645 0.363954 0.000305
3 0.492567 0.243645 0.363954 0.000305
MULLIKEN ATOMIC POPULATION IN EACH
MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 2.000447 -0.000219 -0.000286 1.240897 0.408866
2 -0.000223 1.000109 1.000143 0.379551 0.795567
3 -0.000223 1.000109 1.000143 0.379551 0.795567
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.755336 1.051385 1.304701 0.700792 0.032480
2 0.622332 0.474308 0.347649 0.649604 0.983760
3 0.622332 0.474308 0.347649 0.649604 0.983760
11 12
2.000000 2.000000
1 0.364264 0.000000
2 0.817868 1.000000
3 0.817868 1.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 O 1 S 1.99893 1.99681
2 O 1 S 1.80940 1.73263
3 O 1 X 1.30470 1.29945
4 O 1 Y 1.14192 1.14686
5 O 1 Z 1.60372 1.60700
6 O 2 S 1.99945 1.99865
7 O 2 S 1.92646 1.90037
8 O 2 X 1.34765 1.35028
9 O 2 Y 1.10977 1.15766
10 O 2 Z 1.68733 1.70168
11 O 3 S 1.99945 1.99865
12 O 3 S 1.92646 1.90037
13 O 3 X 1.34765 1.35028
14 O 3 Y 1.10977 1.15766
15 O 3 Z 1.68733 1.70168
----- POPULATIONS IN EACH AO
-----
MULLIKEN LOWDIN
1 O 1 S 1.99893 1.99680
2 O 1 S 1.80783 1.73104
3 O 1 X 1.31391 1.30875
4 O 1 Y 1.14313 1.14789
5 O 1 Z 1.59446 1.59783
6 O 2 S 1.99945 1.99865
7 O 2 S 1.92647 1.90038
8 O 2 X 1.35817 1.36075
9 O 2 Y 1.11032 1.15813
10 O 2 Z 1.67646 1.69094
11 O 3 S 1.99945 1.99865
12 O 3 S 1.92647 1.90038
13 O 3 X 1.35817 1.36075
14 O 3 Y 1.11032 1.15813
15 O 3 Z 1.67646 1.69094
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 O 1 S 1.99892 1.99649
2 O 1 S 1.77896 1.70131
3 O 1 X 1.99929 1.99929
4 O 1 Y 1.08303 1.09222
5 O 1 Z 1.01585 1.02848
6 O 2 S 1.99948 1.99865
7 O 2 S 1.92276 1.89440
8 O 2 X 1.99980 1.99980
9 O 2 Y 0.85538 0.89779
10 O 2 Z 1.28456 1.30048
11 O 3 S 1.99948 1.99865
12 O 3 S 1.92276 1.89440
13 O 3 X 1.99980 1.99980
14 O 3 Y 0.85538 0.89779
15 O 3 Z 1.28456 1.30048
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 O 1 S 1.99893 1.99681
2 O 1 S 1.80940 1.73263
3 O 1 X 1.30470 1.29945
4 O 1 Y 1.14192 1.14686
5 O 1 Z 1.60372 1.60700
6 O 2 S 1.99945 1.99865
7 O 2 S 1.92646 1.90037
8 O 2 X 1.34765 1.35028
9 O 2 Y 1.10977 1.15766
10 O 2 Z 1.68733 1.70168
11 O 3 S 1.99945 1.99865
12 O 3 S 1.92646 1.90037
13 O 3 X 1.34765 1.35028
14 O 3 Y 1.10977 1.15766
15 O 3 Z 1.68733 1.70168
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 7.4593231
2 0.1996707 7.8962121
3 0.1996707 -0.0252150 7.8962121
----- MULLIKEN ATOMIC
OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 7.4600285
2 0.1991142 7.8970318
3 0.1991142 -0.0252744 7.8970318
----- MULLIKEN ATOMIC OVERLAP
POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 7.4593229
2 0.1996708 7.8962120
3 0.1996708 -0.0252150 7.8962120
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 O1 7.858665 0.141335 7.782744 0.217256
2 O2 8.070668 -0.070668 8.108628 -0.108628
3 O2 8.070668 -0.070668 8.108628 -0.108628
TOTAL MULLIKEN AND LOWDIN
ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 O1 7.858257 0.141743 7.782315 0.217685
2 O2 8.070872 -0.070872 8.108843 -0.108843
3 O2 8.070872 -0.070872 8.108843 -0.108843
TOTAL MULLIKEN AND LOWDIN ATOMIC
POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 O1 7.858664 0.141336 7.782744 0.217256
2 O2 8.070668 -0.070668 8.108628 -0.108628
3 O2 8.070668 -0.070668 8.108628 -0.108628
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.278 1.359 1 3 1.278 1.359 2 3 2.177 0.499
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 O1 2.717 2.717 0.000
2 O2 1.858 1.858 0.000
3 O2 1.858 1.858 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 0.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 -0.480768 0.480768
...... END OF PROPERTY EVALUATION ......
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 0.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 -0.491332 0.491332
...... END OF PROPERTY EVALUATION ......
POINT 1 X
Y Z (BOHR) CHARGE
0.000000 0.000000
0.000000 0.00 (A.U.)
DX
DY DZ
/D/ (DEBYE)
0.000000 0.000000
-0.810602 0.810602
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 0.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 -0.480768 0.480768
...... END OF PROPERTY EVALUATION ......
H2
!
! H2-Molekuel, GVB, R=0.7341768A
!
$CONTRL SCFTYP=GVB MULT=1 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=1000 MEMORY=5000000 $END
$BASIS GBASIS=N31 NGAUSS=6 $END
$SCF NCO=0 NSETO=0 NPAIR=1 $END
$GUESS GUESS=HUCKEL $END
$DATA
H2-Molekuel, CI
DNH 2
H 1.0 0.00000 0.00000 0.36709
$END
*************************
ROHF-GVB INPUT PARAMETERS
*************************
NORB = 2 NCO = 0
NPAIR = 1 NSETO = 0
PAIR ORBITALS
PAIR 1 HAS ORBS 1 2
----------------------------
ROHF-GVB COUPLING PARAMETERS
----------------------------
F VECTOR (OCCUPANCIES)
1 0.9529411765
2 0.0470588235
ALPHA COUPLING COEFFICEINTS
1 2
1 0.9529412
2 0.0000000 0.0470588
BETA COUPLING COEFFICIENTS
1 2
1 0.0000000
2 -0.2117647 0.0000000
NATURAL ORBITAL COEFFICIENTS
N.O. PAIR CICOEF-S
1 0.9761870602 -0.2169304578
------------------------
ROHF-GVB SCF CALCULATION
------------------------
GVB STEP WILL USE 50204 WORDS OF MEMORY.
MAXIT= 30 NPUNCH= 2 SQCDF TOL=1.0000E-05
NUCLEAR ENERGY= 0.7207731745
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
ITER EX TOTAL ENERGY E CHANGE SQCDF DIIS ERROR
0 0 -1.141922501 -1.141922501 0.040118149 0.000000000
1 1 -1.145988783 -0.004066282 0.008153342 0.000000000
2 2 -1.146108642 -0.000119860 0.004873220 0.000000000
3 3 -1.146114724 -0.000006082 0.002499592 0.000000000
4 0 -1.146115670 -0.000000946 0.000005395 0.000000000
5 1 -1.146115670 0.000000000 0.000002713 0.000000000
6 2 -1.146115670 0.000000000 0.000001269 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -1.1461156700 AFTER 6 ITERATIONS
Vergleich mit RHF
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 0.7207731745
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=F
DENSITY CONV= 1.00E-05
MEMORY REQUIRED FOR RHF STEP= 7665 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0 -1.120969881 -1.120969881 0.044952909 0.000000000
2 1 0 -1.126681102 -0.005711221 0.006510906 0.000000000
3 2 0 -1.126811882 -0.000130780 0.000963725 0.000000000
4 3 0 -1.126814787 -0.000002905 0.000143169 0.000000000
5 4 0 -1.126814852 -0.000000064 0.000021281 0.000000000
6 5 0 -1.126814853 -0.000000001 0.000003163 0.000000000
7 6 0 -1.126814853 0.000000000 0.000000470 0.000000000
FINAL ENERGY IS -1.1268148531 AFTER 7 ITERATIONS
----------------------------
SCF STATISTICS PER ITERATION
----------------------------
NUMBER OF INTEGRAL PASSES 1
FOCK FORMATION TIME 0.000
GEMINAL OPT TIME 0.000
MIXORB OPT TIME 0.000
OCBSE OPT TIME 0.000
----------------
PAIR INFORMATION
----------------
ORBITAL CI COEFFICIENTS OCCUPATION NUMBERS GVB ENERGY
PAIR 1 2 ORB 1 ORB 2 ORB 1 ORB 2 OVERLAP LOWERING
1 1 2 0.994071 -0.108735 1.97635 0.02365 0.80280 -0.01943
THE MAXIMUM LAGRANGIAN ASYMMETRY IS 0.0000000E+00
------------
EIGENVECTORS
------------
1 2 3 4
-0.6000 -0.0002 0.7742 0.9942
AG B1U AG B1U
1 H 1 S 0.333609 0.772164 0.761241 -0.830511
2 H 1 S 0.265187 0.575313 -0.687898 2.119979
3 H 2 S 0.333609 -0.772164 0.761241 0.830511
4 H 2 S 0.265187 -0.575313 -0.687898 -2.119979
Vergleich mit RHF
FINAL ENERGY IS -1.1268148531 AFTER 7 ITERATIONS
------------
EIGENVECTORS
------------
1 2 3 4
-0.5976 0.2400 0.7719 1.4122
AG B1U AG B1U
1 H 1 S 0.327882 0.121503 0.763726 -1.127486
2 H 1 S 0.270347 1.729745 -0.685887 1.353986
3 H 2 S 0.327882 -0.121503 0.763726 1.127486
4 H 2 S 0.270347 -1.729745 -0.685887 -1.353986
...... END OF RHF CALCULATION ......
-----------
GI ORBITALS
-----------
PAIR 1
1 2
1 H 1 S 0.559198 -0.074274
2 H 1 S 0.432425 -0.071124
3 H 2 S 0.074274 -0.559198
4 H 2 S 0.071124 -0.432425
------------------------------
properties for the GVB density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -2.4864681969
TWO ELECTRON ENERGY = 0.6195793525
NUCLEAR REPULSION ENERGY = 0.7207731745
------------------
TOTAL ENERGY = -1.1461156700
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.6195793525
NUCLEUS-ELECTRON POTENTIAL ENERGY = -3.6607469129
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 0.7207731745
------------------
TOTAL POTENTIAL ENERGY = -2.3203943859
TOTAL KINETIC ENERGY = 1.1742787159
VIRIAL RATIO (V/T) = 1.9760167279
Vergleich mit RHF
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -2.4999950970
TWO ELECTRON ENERGY = 0.6524070694
NUCLEAR REPULSION ENERGY = 0.7207731745
------------------
TOTAL ENERGY = -1.1268148531
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.6524070694
NUCLEUS-ELECTRON POTENTIAL ENERGY = -3.6310514157
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 0.7207731745
------------------
TOTAL POTENTIAL ENERGY = -2.2578711719
TOTAL KINETIC ENERGY = 1.1310563187
VIRIAL RATIO (V/T) = 1.9962499961
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2
1.976354 0.023646
1 0.988177 0.011823
2 0.988177 0.011823
RHF:
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2.000000
1 1.000000
2 1.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.53487 0.50128
2 H 1 S 0.46513 0.49872
3 H 2 S 0.53487 0.50128
4 H 2 S 0.46513 0.49872
RHF
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.52135 0.49009
2 H 1 S 0.47865 0.50991
3 H 2 S 0.52135 0.49009
4 H 2 S 0.47865 0.50991
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2
1 0.6248987
2 0.3751013 0.6248987
RHF
1 2
1 0.5945962
2 0.4054038 0.5945962
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H 1.000000 0.000000 1.000000 0.000000
2 H 1.000000 0.000000 1.000000 0.000000
RHF
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H 1.000000 0.000000 1.000000 0.000000
2 H 1.000000 0.000000 1.000000 0.000000
| Seitenanfang |