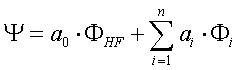
| Input - allgemein |
CI: Configuration Interaction - Theoretischer Hintergrund
Die CI-Methode dient der Berücksichtigung der Elektronenkorrelation. Unter Elektronenkorrelation versteht man, dass die Elektronen versuchen sich auszuweichen, d. h. wenn ein Elektron an einem bestimmten Ort ist, versucht das andere diesen Ort zu meiden (wegen der elektrostatischen Abstoßung und wegen des Pauli-Prinzips). In der HF-Näherung wird die Wechselwirkung eines Elektrons mit den restlichen n-1 Elektronen durch die Wechselwirkung des Elektrons mit einem mittleren Feld, erzeugt durch die restlichen Elektronen, beschrieben. Dieses Feld ist statisch und vernachlässigt die Beeinflussung der Elektronenbewegung durch die Bewegung aller anderen Elektronen bzw. umgekehrt. Die Elektronen weichen einander aus, wodurch die Elektronenabstoßung kleiner als im HF-Modell ist.
Die Gesamtwellenfunktion wird als Multideterminante
dargestellt:
FHF: Slaterdeterminante, Fi:
angeregte Slaterdeterminanten s. folgende Abbildung:
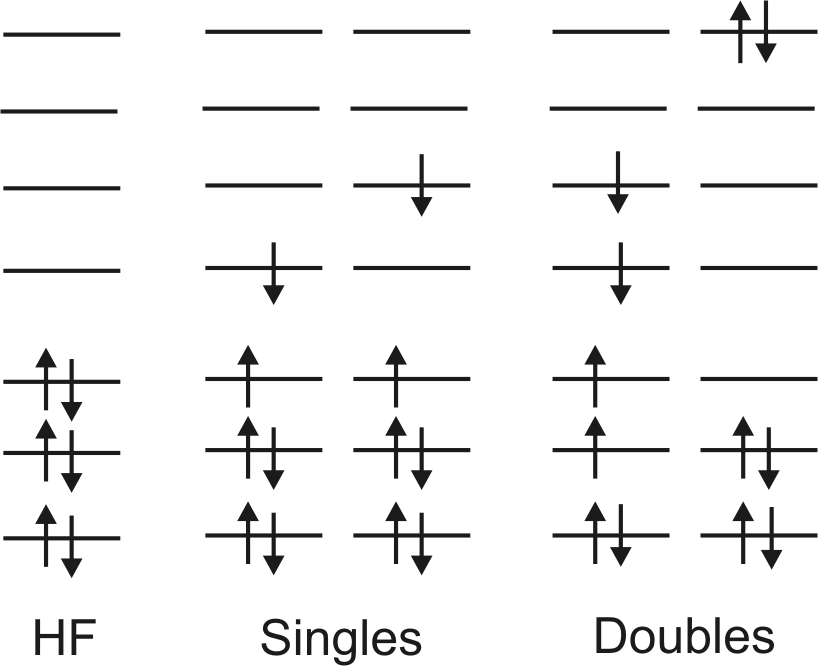
HF: HF-Reference-Konfiguration, Singles: einfach-angeregte Konfigurationen,
Doubles: doppelt-angeregte
Konfigurationen
Die Koeffizienten ai werden so lange variiert, bis die Energie ein Minimum ist. Die MO's (F's) sind die HF-MO's und bleiben unverändert (bei MCSCF werden auch diese variiert).
Diese Variationsrechnung bedeutet Aufstellung der CI-Matrix
und Diagonalisierung der CI-Matrix. Die dann erhaltenen Eigenwerte stellen die
CI-Energie und die Eigenvektoren die Koeffizienten ai dar.
Die Größe der CI-Matrix ergibt sich aus den Kombinationsmöglichkeiten der
Elektronen aus besetzten MO's in unbesetzte MO's. Sie wächst dramatisch mit der
Größe des Basissatzes und der angeregten Elektronen (IEXCIT). Allerdings sind
die meisten Matrixelemente aus verschiedenen Gründen (Symmetrie,
Orthogonalität) gleich Null. Man sucht deshalb nach Diagonalisierungsverfahren,
die es gestatten nur wenige Eigenwerte und Eigenvektoren zu extrahieren (anstatt
alle zu berechnen). Bei einer CI-Rechnung mit CITYP=GUGA geschieht dies mit dem
Davidson-Algorithmus. Dabei wird mit einem
GUESS-Eigenvektor gestartet, der
durch WSTATE definiert wird.
In den folgenden Beispielen kann man Details einer solchen CI-Rechnung ansehen: H2,
Diese Beschreibung der CI-Methode suggeriert, dass bei Ein-Elektronensystemen CI keinen Effekt haben sollte. Dem ist aber nicht so: H-Atom, H2+.
Bei anderen 1-Elektronensystemen funktioniert das aber aus irgendwelchen Gründen nicht: lineares H43+,
Führt man eine Berechnung des H-Atoms mit dem Basissatz STO-6-311G NPFUNC=3 und DIFFS durch,
Input-File:
!
! H atom
!
$CONTRL SCFTYP=ROHF MULT=2 RUNTYP=ENERGY $END
$SYSTEM TIMLIM=1000 MEMORY=5000000 $END
$BASIS GBASIS=N311 NGAUSS=6 NPFUNC=3 DIFFS=.TRUE. $END
$GUESS GUESS=HUCKEL $END
$DATA
H-Atom
C1
H 1.0 0.00000 0.00000 0.00000
$END
so erhält man folgende Atom-Orbitale (nur die ersten fünf von 13 Orbitalen sind hier angegeben):
------------
EIGENVECTORS
------------
1 2 3 4 5
-0.4998 0.0792 0.3629 0.3629 0.3629
A A A A A
1 H 1 S 0.237599 -0.084489 0.000000 0.000000 0.000000
2 H 1 S 0.506406 0.012956 0.000000 0.000000 0.000000
3 H 1 S 0.375218 -1.076603 0.000000 0.000000 0.000000
4 H 1 S 0.008363 1.659519 0.000000 0.000000 0.000000
5 H 1 X 0.000000 0.000000 0.009778 -0.009483 0.017047
6 H 1 Y 0.000000 0.000000 0.016473 -0.006198 -0.012897
7 H 1 Z 0.000000 0.000000 0.010447 0.018649 0.004382
8 H 1 X 0.000000 0.000000 -0.034538 0.033496 -0.060213
9 H 1 Y 0.000000 0.000000 -0.058188 0.021894 0.045556
10 H 1 Z 0.000000 0.000000 -0.036903 -0.065872 -0.015477
11 H 1 X 0.000000 0.000000 0.465764 -0.451710 0.812007
12 H 1 Y 0.000000 0.000000 0.784693 -0.295253 -0.614342
13 H 1 Z 0.000000 0.000000 0.497649 0.888323 0.208714
Als Energie des Grundzustandes würde man die Energie des ersten AO’s, für die des ersten angeregten Zustandes die des zweiten AO’s usw. angeben.
Führt man jetzt eine CI-Rechnung durch,
Input-File:
!
! H atom, ci, 5 electronic states
!
!
$CONTRL SCFTYP=ROHF MULT=2 RUNTYP=ENERGY CITYP=GUGA $END
$SYSTEM TIMLIM=1000 MEMORY=5000000 $END
$BASIS GBASIS=N311 NGAUSS=6 NPFUNC=3 DIFFS=.TRUE. $END
$CIDRT GROUP=C1 IEXCIT=1 NFZC=0 NDOC=0 NALP=1 NVAL=12 $END
$GUGDIA NSTATE=5 $END
$GUGDM IROOT=1 NFLGDM(1)=1 $END
$GUESS GUESS=HUCKEL $END
$DATA
H-Atom, ci, 5 states
C1
H 1.0 0.00000 0.00000 0.00000
$END
so erhält man folgendes Resultat:
STATE # 1 ENERGY = -0.499817916
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
1 1.000000 1000000000000
STATE # 2 ENERGY = -0.118768036
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
9 0.171967 0000010000000
13 0.985089 0100000000000
STATE # 3 ENERGY = 0.006725206
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
10 -0.520067 0000100000000
11 0.839166 0001000000000
12 0.153987 0010000000000
STATE # 4 ENERGY = 0.006725206
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
10 0.107934 0000100000000
11 -0.114179 0001000000000
12 0.986760 0010000000000
STATE # 5 ENERGY = 0.006725206
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
10 0.846322 0000100000000
11 0.530231 0001000000000
...... END OF CI-MATRIX DIAGONALIZATION ......
Die Energie der angeregten Zustände, vor allem des ersten angeregten Zustandes, kommt jetzt wesentlich besser heraus.
Versteht man unter CI einfach eine Erweiterung des Orbitalraumes [1], kann man dieses Ergebnis möglicherweise verstehen.