Hartree-Fock self-consistent field (SCF) method
- beginn with a set of approximate orbitals for all the electrons in the
system (e.g., GUESS=HUCKEL)
- one electron is selected, and the potential in which it moves is
calculated by freezing the distribution of all the other electrons and
treating their averaged distribution as the centrosymmetric source of
potential
- the Schroedinger equation is solved for this potential, which gives a new
potential for it
- the procedure is repeated for all the other electrons in the system, using
the electrons in the frozen orbitals as source of the potential
- at the end of one cycle, there are new
orbitals from the original set
- the process is repeated until there is little or no change in the orbitals
Hartree Fock method
-
Abbreviation HF or RHF
-
Simplest method, closely related to what we teach in the stage 1 MO course
-
Good enough for most geometry calculations
-
May be good enough for reaction energetics
-
Just as geometry optimisations work by minimising energy with respect to
geometry, so ab initio calculations, for a single geometry
point, work by minimising the electronic energy by changing the array
of coefficients which control how AOs are added together to make each MO
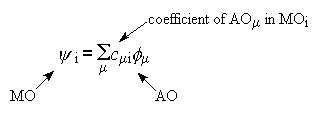
-
The Variation Principle says that if there are two approximations to a
wavefunction (i.e. two different sets of coefficients c), then the
better approximation is the one which gives the lower energy
-
The HF method works by calculating orbitals for one electron in an average
field of the rest, as follows:
-
Start with guess at the electron distribution
-
At the start of a calculation, this comes from a semiempirical calculation
-
Use the guess to calculate the average field of the rest of the electrons
-
Calculate coefficients to produce a set of one-electron MOs and energies
-
Fill the MOs according to the Aufbau principle and get total electronic
energy
-
Use this set of MOs as the next approximation to the electron distribution
-
Repeat the process with gradually improving MOs, until the electronic energy
converges, i.e. it changes by less than some pre-set limit
-
This is called self consistent field (SCF) convergence
-
If we are doing a geometry optimisation, the program moves on to the next
geometry point
-
The AOs move with the atoms
-
The set of coefficients from the last geometry point can be a starting
point for the new SCF calculation, and so on, until the geometry optimisation
converges
-
Because we have one iterative process, the SCF minimisation, inside another
iterative process, the geometry minimisation, the whole process takes a
lot longer than force-field or semi-empirical methods
-
The program can save a lot of time if it knows that all electrons are paired,
so it can treat them two at a time
-
This is so for ordinary, diamagnetic organic compounds, for most main-groups
compounds, and for organometallic compounds which obey the 18-electron
rule
-
This is called a restricted Hartree Fock treatment (RHF)
-
Treating compounds with unpaired electrons is much more difficult (ROHF,
UHF)
UHF berücksichtigt in gewisser Weise die Elektronenkorrelation (CI ist mit
UHF auch gar nicht möglich).