HINT (Hilderbrandt
INTernal Coordinates)
Diese Art von internen Koordinaten ist extrem schwierig zu
verstehen. Es nutzt dabei auch nicht viel, die empfohlene Literaturstelle zu
lesen (R. L. Hilderbrandt, Cartesian Coordinates of Molecular Models, J. Chem.
Phys. 51, 1654 (1969)). Da die nichtanalytische Geometrieoptimierung angeregter
Zustände nur mit diesen Koordinaten funktioniert, ist es aber notwendig, damit
arbeiten zu können. Ich versuche es anhand einiger Beispiele.
COORD=HINT: only symmetry unique atoms will be given, in
Hilderbrandt style internals
!
! H2O
!
$CONTRL SCFTYP=RHF MULT=1 RUNTYP=TRUDGE CITYP=GUGA COORD=HINT $END
$SYSTEM TIMLIM=100 MEMORY=5000000 $END
$BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 $END
$GUESS GUESS=HUCKEL $END
$TRUDGE OPTMIZ=GEOMETRY NPAR=2 IEX(1)=21,22 P(1)=0.96 $END
$CIDRT GROUP=C2V SOCI=.TRUE. NFZC=1 NDOC=4 NVAL=14 NEXT=-1 $END
$DATA
Water CI-SD Geometry Optimization
CNV 2
O 8.0 LC 0.0
0.0 0.0 - O K
H 1.0 PCC 0.94 53.0
0.0 + O K I
$END
ändert man die Koordinatenzeilen wie
folgt:
O 8.0 LC 0.0
0.0 0.0 - O
H 1.0 PCC 0.94 53.0
0.0
+ O K
kommt aber das Gleiche heraus.
NAME, ZNUC, CONX, R, ALPHA, BETA, SIGN,
POINT1, POINT2, POINT3
NAME: 10 character atomic name (used
only for print out).
ZNUC: nuclear charge.
CONX: connection type, choose from:
LC: linear connection: 0.0: O-Atom ist am Koordinaten-Ursprung;
keine Winkel
PCC: planar central
connection: 0.94: H-Atom ist 0.94 A vom O-Atom entfernt und bildet mit der
Drehachse
einen Winkel von 53°, 0.0: die eingeschlossene Fläche ist nicht verdrillt?
NPCC: non-planar central
connection
PTC : planar terminal
connection
CCPA : central connection
with polar atom
TCT: terminal connection
with torsion
R: connection distance
ALPHA: first connection angle
BETA: second connection angle
SIGN: connection sign “+” or “-“:
spielt im obigen Fall keine Rolle.
POINT1, POINT2, POINT3: connection
points, a serial number of a previously input atom, or one of 4 standard points:
O, I, J, K (origin and unit points on axes of master frame). Default: POINT1=O,
POINT2=I, POINT3=J
Am besten geht es durch probieren. Z. B. das N2O4-Molekül
!
! N2O4-Molekuel
!
!
$CONTRL SCFTYP=RHF MULT=1 RUNTYP=OPTIMIZE COORD=HINT $END
$SYSTEM TIMLIM=1000 MEMORY=5000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
N2O4-Molekuel
CNH 2
N 7.0 LC 0.90 0.0
0.0 - O
O 8.0 PCC 1.20 112.0 0.0 + 1
O 8.0 PCC 1.20
-112.0 0.0 +
1
$END
ergibt:
ATOM ATOMIC COORDINATES (BOHR)
CHARGE
X
Y
Z
N
7.0 -1.7007533889 0.0000000000 0.0000000000
N
7.0 1.7007533889 0.0000000000 0.0000000000
O
8.0 -0.8512688112 -2.1025481104 0.0000000000
O
8.0 0.8512688112
2.1025481104 0.0000000000
O
8.0 -0.8512688112
2.1025481104 0.0000000000
O
8.0 0.8512688112 -2.1025481104 0.0000000000
Man sieht, dass die beiden N-Atome auf der x-Achse liegen,
die O-Atome liegen aber näher zum Ursprung, d. h. folgende Struktur liegt vor:
O O
N
N
O O
Man muss also die Eingabe ändern.
N 7.0 LC 0.90 0.0 0.0 + O
O 8.0 PCC 1.20 112.0 0.0 + 1
O 8.0 PCC 1.20 -112.0 0.0 + 1
Das ist die Lösung:
N 7.0 1.7007533889 0.0000000000 0.0000000000
N 7.0 -1.7007533889 0.0000000000 0.0000000000
O 8.0 2.5502379666 -2.1025481104 0.0000000000
O 8.0 -2.5502379666 2.1025481104 0.0000000000
O 8.0 2.5502379666 2.1025481104 0.0000000000
O 8.0 -2.5502379666 -2.1025481104 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
N N O O
1 N 0.0000000 1.8000000 * 1.2000000 * 2.5096415 *
2 N 1.8000000 * 0.0000000 2.5096415 * 1.2000000 *
3 O 1.2000000 * 2.5096415 * 0.0000000 3.4980853
4 O 2.5096415 * 1.2000000 * 3.4980853 0.0000000
5 O 1.2000000 * 2.5096415 * 2.2252413 * 2.6990558 *
6 O 2.5096415 * 1.2000000 * 2.6990558 * 2.2252413 *
Nach der Geometrieoptimierung:
***** EQUILIBRIUM GEOMETRY LOCATED *****
N2O4-Molekuel
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X
Y
Z
------------------------------------------------------------
N 7.0
1.4182942123 0.0000000002
0.0000000000
O 8.0
0.6801493554 1.1710604612
0.0000000000
O 8.0
0.6801493560 -1.1710604626
0.0000000000
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
N 7.0 -1.4182942123 -0.0000000002 0.0000000000
N 7.0 1.4182942123 0.0000000002 0.0000000000
O 8.0 -0.6801493554 -1.1710604612 0.0000000000
O 8.0 0.6801493554 1.1710604612 0.0000000000
O 8.0 -0.6801493560 1.1710604626 0.0000000000
O 8.0 0.6801493560 -1.1710604626 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
N N O O
1 N 0.0000000 2.8365884 * 1.3842834 * 2.4030913 *
2 N 2.8365884 * 0.0000000 2.4030913 * 1.3842834 *
3 O 1.3842834 * 2.4030913 * 0.0000000 2.7084946 *
4 O 2.4030913 * 1.3842834 * 2.7084946 * 0.0000000
5 O 1.3842834 * 2.4030913 * 2.3421209 * 1.3602987 *
6 O 2.4030913 * 1.3842834 * 1.3602987 * 2.3421209 *
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
N
N
O
O
1 N 0.0000000
1.8000000 * 1.2000000
* 1.7497713 *
2 N 1.8000000 *
0.0000000 1.7497713
* 1.2000000 *
3 O 1.2000000 *
1.7497713 * 0.0000000
2.4007080 *
4 O 1.7497713 *
1.2000000 * 2.4007080
* 0.0000000
5 O 1.2000000 *
1.7497713 * 2.2252413
* 0.9009442 *
6 O 1.7497713 *
1.2000000 * 0.9009442
* 2.2252413 *
* ... LESS THAN 3.000
Obige Logik funktioniert aber nicht, wenn
im Koordinatenursprung ein Atom steht, z. B. NO2:
N 7.0 LC
0.00 0.0 0.0 + O
O 8.0 PCC 1.20 56.0 0.0
+ 1
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
N 7.0 0.0000000000 0.0000000000
0.0000000000
O 8.0 -1.2680656342 -1.8799846148
0.0000000000
O 8.0 1.2680656342 -1.8799846148
0.0000000000
O 8.0 -1.2680656342 1.8799846148
0.0000000000
O 8.0 1.2680656342 1.8799846148
0.0000000000
Man muss wie oben im ersten Beispiel
(H2O)
verfahren:
N 7.0 LC
0.00 0.0 0.0 - O K
O 8.0 PCC
1.20 56.0 0.0 + O K I
und erhält dann:
ATOM ATOMIC
COORDINATES (BOHR)
CHARGE X
Y
Z
N 7.0 0.0000000000 0.0000000000
0.0000000000
O 8.0 -1.8799846148 0.0000000000
1.2680656342
O 8.0 1.8799846148 0.0000000000
1.2680656342
Bicyclopropylium
Bicyclopropylium-Molekuel
CNH 2
C 6.0 LC 0.75 0.0 0.0 + O
C 6.0 PCC 1.40 150.0 0.0 + 1
C 6.0 PCC 1.40 -150.0 0.0 + 1
H 1.0 LC 1.00 0.0 0.0 + 2
H 1.0 LC 1.00 0.0 0.0 + 3
ergibt:
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
C 6.0 1.4172944908 0.0000000000 0.0000000000
C 6.0 -1.4172944908 0.0000000000 0.0000000000
C 6.0 3.7084654870 -1.3228081914 0.0000000000
C 6.0 -3.7084654870 1.3228081914 0.0000000000
C 6.0 3.7084654870 1.3228081914 0.0000000000
C 6.0 -3.7084654870 -1.3228081914 0.0000000000
H 1.0 5.5475472041 -1.7573685974 0.0000000000
H 1.0 -5.5475472041 1.7573685974 0.0000000000
H 1.0 5.5475472041 1.7573685974 0.0000000000
H 1.0 -5.5475472041 -1.7573685974 0.0000000000
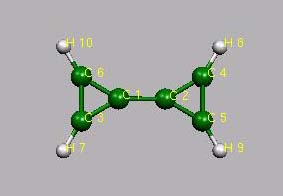 Bild
ist allerdings verdreht!
Bild
ist allerdings verdreht!
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
C C C C
1 C 0.0000000 1.5000000 * 1.4000000 * 2.8013045 *
2 C 1.5000000 * 0.0000000 2.8013045 * 1.4000000 *
3 C 1.4000000 * 2.8013045 * 0.0000000 4.1670869
4 C 2.8013045 * 1.4000000 * 4.1670869 0.0000000
5 C 1.4000000 * 2.8013045 * 1.4000000 * 3.9248711
6 C 2.8013045 * 1.4000000 * 3.9248711 1.4000000 *
7 H 2.3752533 * 3.8011493 1.0000000 * 5.1621576
8 H 3.8011493 2.3752533 * 5.1621576 1.0000000 *
9 H 2.3752533 * 3.8011493 1.8983905 * 4.9034665
10 H 3.8011493 2.3752533 * 4.9034665 1.8983905 *
$TRUDGE: non-gradient optimization
NPAR= number of parametres to be
optimized
IEX= defines the parameters to be
optimized. Pointers to the HINT internal coordinates which will be optimized.
The pointers to the internal coordinates are defined as: (the number of atom in
the input list)*10 + (the number of internal coordinate for that atom). For
each atom, the HINT internal coordinates are numbered as 1, 2, and 3 for BOND,
ALPHA, and BETA, respectively.
P= Defines the initial values of the
parameters to be optimized. If omitted, the $DATA values are used.
-----------------------------------------------------------
INTERNAL COORDINATES (ANGSTROMS/DEGREE)
I BOND ALPHA BETA SIGN ICONX IATCON INATOM
1 0.0000000 0.00000 0.00000 -1.0 1501504503 1
2 0.9600000 53.00000 0.00000 1.0 2501504502 3
-----------------------------------------------------------
----- TRUDGE RESTART DATA AT NSTEP 0 --------
$TRUDGE OPTIMIZE=GEOMETRY NPAR= 2
IEX(1)= 21, 22,
P(1)= 0.960000, 53.000000,
$END
TRUDGE ENERGY VALUE AT NSTEP= 0 IS -76.0103894679
-----------------------------------------------------------
INTERNAL COORDINATES (ANGSTROMS/DEGREE)
I BOND ALPHA BETA SIGN ICONX IATCON INATOM
1 0.0000000 0.00000 0.00000 -1.0 1501504503 1
2 0.9600000 58.57406 0.00000 1.0 2501504502 3
-----------------------------------------------------------
----- TRUDGE RESTART DATA AT NSTEP 1 --------
$TRUDGE OPTIMIZE=GEOMETRY NPAR= 2
IEX(1)= 21, 22,
P(1)= 0.960000, 58.574059,
$END
TRUDGE ENERGY VALUE AT NSTEP= 1 IS -76.0064779202
LOOKS = 1 ALF = 0.1000 FUNC = -76.006477920 AMIN = 0.0000 FMIN =
-76.010389468 ANEXT = 0.1000 FNEXT = -76.006477920
-----------------------------------------------------------
INTERNAL COORDINATES (ANGSTROMS/DEGREE)
I BOND ALPHA BETA SIGN ICONX IATCON INATOM
1 0.0000000 0.00000 0.00000 -1.0 1501504503 1
2 0.9600000 47.95638 0.00000 1.0 2501504502 3
-----------------------------------------------------------
----- TRUDGE RESTART DATA AT NSTEP 2 --------
$TRUDGE OPTIMIZE=GEOMETRY NPAR= 2
IEX(1)= 21, 22,
P(1)= 0.960000, 47.956383,
$END
TRUDGE ENERGY VALUE AT NSTEP= 2 IS -76.0078393859
LOOKS = 2 ALF = -0.1000 FUNC = -76.007839386 AMIN = 0.0000 FMIN =
-76.010389468 ANEXT = -0.1000 FNEXT = -76.007839386
-----------------------------------------------------------
INTERNAL COORDINATES (ANGSTROMS/DEGREE)
I BOND ALPHA BETA SIGN ICONX IATCON INATOM
1 0.0000000 0.00000 0.00000 -1.0 1501504503 1
2 0.9600000 52.44458 0.00000 1.0 2501504502 3
-----------------------------------------------------------
----- TRUDGE RESTART DATA AT NSTEP 3 --------
$TRUDGE OPTIMIZE=GEOMETRY NPAR= 2
IEX(1)= 21, 22,
P(1)= 0.960000, 52.444576,
$END
TRUDGE ENERGY VALUE AT NSTEP= 16 IS -76.0107457254
NON-GRADIENT ENERGY MINIMIZATION ... CONVERGED
CONJUGATE DIRECTION = 0
PARAMETER = 1
VALUE OF THE FUNCTION = -76.0107457254
FNOT = -76.0107457254
$TRURST KSTART= 0 JSTART= 1 TOLF= 0.001000 TOLR= 0.050000
FNOISE= 0.000500 FNOT= -76.010745725
RNOT(1)= 0.007667 -0.006251
CURV(1)= 1.901800 0.321617
ALPH(1)= 0.012500 0.012500
V(1)= 1.000000 0.000000
0.000000 1.000000
$END
-----------------------------------------------------------
INTERNAL COORDINATES (ANGSTROMS/DEGREE)
I BOND ALPHA BETA SIGN ICONX IATCON INATOM
1 0.0000000 0.00000 0.00000 -1.0 1501504503 1
2 0.9472348 52.66973 0.00000 1.0 2501504502 3
-----------------------------------------------------------
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
O 8.0 0.0000000000 0.0000000000 0.0000000000
H 1.0 -0.7531968076 0.0000000000 0.5744113322
H 1.0 0.7531968076 0.0000000000 0.5744113322
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
O H H
1 O 0.0000000 0.9472348 * 0.9472348 *
2 H 0.9472348 * 0.0000000 1.5063936 *
3 H 0.9472348 * 1.5063936 * 0.0000000
Benzol
!
! Benzol, HINT
!
$CONTRL SCFTYP=RHF MULT=1 RUNTYP=TRUDGE
COORD=HINT $END
$TRUDGE OPTMIZ=GEOMETRY NPAR=2 IEX(1)=11,21 $END
$SYSTEM TIMLIM=10 MEMORY=6000000 $END
$BASIS GBASIS=N31 NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
Benzene
Dnh 6
C 6.0 LC 1.390 0.0 0.0 + O
H 1.0 LC 2.470 0.0 0.0 + O
$END
Man erkennt, dass sich Linear Connection (LC) auf den
Abstand zum Ursprung (O, Origin) bezieht. Das Vorzeichen spielt hier keine
Rolle.