Cyclic H3+
6G, Singulett
Input
!
! cycl H3_1plus_singl
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=1
RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=60000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
cycl H3_2plus S0
CNH 3
H 1.0 0.00000 1.000 0.00000
$END
Output
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
H 1.0 -1.6365507116
-0.9448629939 0.0000000000
H 1.0 1.6365507116
-0.9448629939 0.0000000000
H 1.0 0.0000000000
1.8897259877 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
H
H H
1 H 0.0000000 1.7320508 *
1.7320508 *
2 H 1.7320508 * 0.0000000
1.7320508 *
3 H 1.7320508 * 1.7320508 *
0.0000000
ATOMIC BASIS SET
----------------
THE CONTRACTED PRIMITIVE FUNCTIONS HAVE BEEN UNNORMALIZED
THE CONTRACTED BASIS FUNCTIONS ARE NOW NORMALIZED TO UNITY
SHELL TYPE PRIM EXPONENT CONTRACTION COEFFICIENTS
H
3 S 1 35.523221 0.095030 ( 0.009164)
3 S 2 6.513144 0.143430 ( 0.049361)
3 S 3 1.822143 0.188385 ( 0.168538)
3 S 4 0.625955 0.185857 ( 0.370563)
3 S 5 0.243077 0.102760 ( 0.416492)
3 S 6 0.100112 0.016532 ( 0.130334)
TOTAL NUMBER OF SHELLS = 3
TOTAL NUMBER OF BASIS FUNCTIONS = 3
NUMBER OF ELECTRONS = 2
CHARGE OF MOLECULE = 1
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 1
NUMBER OF OCCUPIED ORBITALS (BETA ) = 1
TOTAL NUMBER OF ATOMS = 3
THE NUCLEAR REPULSION ENERGY IS 0.9165618819
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 0.9165618819
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=F
DENSITY CONV= 1.00E-05
MEMORY REQUIRED FOR RHF STEP= 7602 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0 -1.046338566 -1.046338566 0.000000000 0.000000000
2 1 0 -1.046338566 0.000000000 0.000000000 0.000000000
-----------------
DENSITY CONVERGED
-----------------
------------
EIGENVECTORS
------------
1 2
3
-0.7540
-0.2213 -0.2213
A' E'
E'
1 H 1 S 0.494268 -0.781927 -0.451446
2 H 2 S 0.494268 0.781927 -0.451446
3 H 3 S 0.494268 0.000000
0.902892
MO1
MO2
MO3



-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -2.4178641130
TWO ELECTRON ENERGY = 0.4549636649
NUCLEAR REPULSION ENERGY = 0.9165618819
------------------
TOTAL ENERGY = -1.0463385662
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.4549636649
NUCLEUS-ELECTRON POTENTIAL ENERGY = -3.5276738711
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 0.9165618819
------------------
TOTAL POTENTIAL ENERGY = -2.1561483243
TOTAL KINETIC ENERGY = 1.1098097581
VIRIAL RATIO (V/T) = 1.9428089441
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2.000000
1 0.666667
2 0.666667
3 0.666667
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.66667 0.66667
2 H 2 S 0.66667 0.66667
3 H 3 S 0.66667 0.66667
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1
2 3
1 0.4886021
2 0.0890323 0.4886021
3 0.0890323 0.0890323 0.4886021
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE
LOW.POP. CHARGE
1 H 0.666667 0.333333
0.666667 0.333333
2 H 0.666667 0.333333
0.666667 0.333333
3 H 0.666667 0.333333
0.666667 0.333333

-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND
BOND
BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST
ORDER
1 2 1.732 0.444 1 3
1.732 0.444 2 3 1.732 0.444
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.889 0.889 0.000
2 H 0.889 0.889 0.000
3 H 0.889 0.889 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 1.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 0.000000 0.000000
...... END OF PROPERTY EVALUATION ......
Triplettzustand
!
! cycl H3_1plus_tripl
!
$CONTRL SCFTYP=ROHF MULT=3 ICHARG=1
RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=60000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
cycl H3_2plus T1
CNH 3
H 1.0 0.00000 1.000 0.00000
$END
Output
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
H 1.0 -1.6365507116 -0.9448629939 0.0000000000
H 1.0 1.6365507116 -0.9448629939 0.0000000000
H 1.0 0.0000000000 1.8897259877 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
H H H
1 H 0.0000000 1.7320508 * 1.7320508 *
2 H 1.7320508 * 0.0000000 1.7320508 *
3 H 1.7320508 * 1.7320508 * 0.0000000
ATOMIC BASIS SET
----------------
THE CONTRACTED PRIMITIVE FUNCTIONS HAVE BEEN UNNORMALIZED
THE CONTRACTED BASIS FUNCTIONS ARE NOW NORMALIZED TO UNITY
SHELL TYPE PRIM EXPONENT CONTRACTION COEFFICIENTS
H
3 S 1 35.523221 0.095030 ( 0.009164)
3 S 2 6.513144 0.143430 ( 0.049361)
3 S 3 1.822143 0.188385 ( 0.168538)
3 S 4 0.625955 0.185857 ( 0.370563)
3 S 5 0.243077 0.102760 ( 0.416492)
3 S 6 0.100112 0.016532 ( 0.130334)
TOTAL NUMBER OF SHELLS = 3
TOTAL NUMBER OF BASIS FUNCTIONS = 3
NUMBER OF ELECTRONS = 2
CHARGE OF MOLECULE = 1
STATE MULTIPLICITY = 3
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 2
NUMBER OF OCCUPIED ORBITALS (BETA ) = 0
TOTAL NUMBER OF ATOMS = 3
THE NUCLEAR REPULSION ENERGY IS 0.9165618819
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 -1.003207884 -1.003207884 0.815213623 0.000000000
2 1 -0.793340598 0.209867286 0.000000000 0.000000000
3 2 -0.793340598 0.000000000 0.000000000 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -0.7933405979 AFTER 3 ITERATIONS
--------------------
SPIN SZ = 1.000
S-SQUARED = 2.000
--------------------
------------
EIGENVECTORS
------------
1 2
3
-0.7026
-0.5337 -0.5337
A' E'
E'
1 H 1 S 0.494268 -0.451446 -0.781927
2 H 2 S 0.494268 -0.451446 0.781927
3 H 3 S 0.494268 0.902892
0.000000
MO1
MO2
MO3



...... END OF ROHF CALCULATION ......
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -2.1835003629
TWO ELECTRON ENERGY = 0.4735978831
NUCLEAR REPULSION ENERGY = 0.9165618819
------------------
TOTAL ENERGY = -0.7933405979
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.4735978831
NUCLEUS-ELECTRON POTENTIAL ENERGY = -3.6851334763
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 0.9165618819
------------------
TOTAL POTENTIAL ENERGY = -2.2949737113
TOTAL KINETIC ENERGY = 1.5016331134
VIRIAL RATIO (V/T) = 1.5283185292
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2
1.000000 1.000000
1 0.333333 0.166667
2 0.333333 0.166667
3 0.333333 0.666667
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H 0.500000 0.500000
2 H 0.500000 0.500000
3 H 1.000000 1.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.50000 0.50000
2 H 2 S 0.50000 0.50000
3 H 3 S 1.00000 1.00000
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 0.4481044
2 0.0816529 0.4481044
3 -0.0297573 -0.0297573 1.0595147
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE
LOW.POP. CHARGE
1 H 0.500000 0.500000
0.500000 0.500000
2 H 0.500000 0.500000
0.500000 0.500000
3 H 1.000000 0.000000
1.000000 0.000000

Die Ladung +1 verteilt sich auf die beiden unteren
H-Atome. Das obere H-Atom trägt die Ladung Null!
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND
ATOM PAIR DIST ORDER
1 2 1.732 0.500
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.750 0.500 0.250
2 H 0.750 0.500 0.250
3 H 1.000 0.000 1.000
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H 1.0 0.2459386
2 H 1.0 0.2459386
3 H 1.0 0.5615829
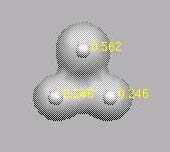
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X
Y Z (BOHR) CHARGE
0.000000 0.000000
0.000000 1.00 (A.U.)
DX
DY DZ
/D/ (DEBYE)
0.000000
-2.401621 0.000000
2.401621
cyclisches H3+ im Triplettzustand hat ein Dipolmoment!
...... END OF PROPERTY EVALUATION ......
Basissatz: 31G
Singulett
!
! cycl H3_1plus_singl
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=1 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=60000000 $END
$BASIS GBASIS=N31 NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
cycl H3_2plus S0
CNH 3
H 1.0 0.00000 1.000 0.00000
$END
SHELL TYPE PRIM EXPONENT CONTRACTION COEFFICIENTS
H
5 S 1 18.731137 0.214935 ( 0.033495)
5 S 2 2.825394 0.364571 ( 0.234727)
5 S 3 0.640122 0.415051 ( 0.813757)
6 S 4 0.161278 0.181381 ( 1.000000)
TOTAL NUMBER OF SHELLS = 6
TOTAL NUMBER OF BASIS FUNCTIONS = 6
NUMBER OF ELECTRONS = 2
CHARGE OF MOLECULE = 1
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 1
NUMBER OF OCCUPIED ORBITALS (BETA ) = 1
TOTAL NUMBER OF ATOMS = 3
THE NUCLEAR REPULSION ENERGY IS 0.9165618819
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 0.9165618819
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=F
DENSITY CONV= 1.00E-05
MEMORY REQUIRED FOR RHF STEP= 7833 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0 -1.025089173 -1.025089173 0.116921507 0.000000000
2 1 0 -1.095888724 -0.070799551 0.015036341 0.000000000
3 2 0 -1.097745036 -0.001856312 0.002376118 0.000000000
4 3 0 -1.097795740 -0.000050704 0.000390869 0.000000000
5 0 0 -1.097797134 -0.000001393 0.000077677 0.000000000
6 1 0 -1.097797173 -0.000000039 0.000000001 0.000000000
7 2 0 -1.097797173 0.000000000 0.000000000 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -1.0977971729 AFTER 7 ITERATIONS
------------
EIGENVECTORS
------------
1 2 3 4 5
-0.7986 -0.3101 -0.3101 0.6649 0.6649
A' E' E' E' E'
1 H 1 S 0.203517 -0.155725 -0.269724 0.527634 0.913889
2 H 1 S 0.302903 -0.381038 -0.659977 -0.605172 -1.048189
3 H 2 S 0.203517 -0.155725 0.269724 0.527634 -0.913889
4 H 2 S 0.302903 -0.381038 0.659977 -0.605172 1.048189
5 H 3 S 0.203517 0.311450 0.000000 -1.055269 0.000000
6 H 3 S 0.302903 0.762076 0.000000 1.210344 0.000000
6
0.7516
A'
1 H 1 S 0.772195
2 H 1 S -0.519425
3 H 2 S 0.772195
4 H 2 S -0.519425
5 H 3 S 0.772195
6 H 3 S -0.519425
...... END OF RHF CALCULATION ......
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -2.4314671829
TWO ELECTRON ENERGY = 0.4171081281
NUCLEAR REPULSION ENERGY = 0.9165618819
------------------
TOTAL ENERGY = -1.0977971729
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.4171081281
NUCLEUS-ELECTRON POTENTIAL ENERGY = -3.1417702330
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 0.9165618819
------------------
TOTAL POTENTIAL ENERGY = -1.8081002230
TOTAL KINETIC ENERGY = 0.7103030500
VIRIAL RATIO (V/T) = 2.5455335196
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2.000000
1 0.666667
2 0.666667
3 0.666667
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.20751 0.20759
2 H 1 S 0.45915 0.45908
3 H 2 S 0.20751 0.20759
4 H 2 S 0.45915 0.45908
5 H 3 S 0.20751 0.20759
6 H 3 S 0.45915 0.45908
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 0.4286626
2 0.1190020 0.4286626
3 0.1190020 0.1190020 0.4286626
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H 0.666667 0.333333 0.666667 0.333333
2 H 0.666667 0.333333 0.666667 0.333333
3 H 0.666667 0.333333 0.666667 0.333333
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.732 0.444 1 3 1.732 0.444 2 3 1.732 0.444
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.889 0.889 0.000
2 H 0.889 0.889 0.000
3 H 0.889 0.889 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 1.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 0.000000 0.000000
...... END OF PROPERTY EVALUATION ......
Triplett
Input
!
! cycl H3_1plus_tripl
!
$CONTRL SCFTYP=ROHF MULT=3 ICHARG=1 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=60000000 $END
$BASIS GBASIS=N31 NGAUSS=6 $END
$GUESS GUESS=MOREAD NORB=6 $END
$DATA
cycl H3_2plus T1
CNH 3
H 1.0 0.00000 1.000 0.00000
$END
--- RHF ORBITALS --- GENERATED AT 16:23:51 LT 4-DEC-2002
cycl H3_2plus S0
E(RHF)= -1.0977971729, E(NUC)= 0.9165618819, 7 ITERS
$VEC
1 1 2.03516751E-01 3.02903099E-01 2.03516751E-01 3.02903099E-01 2.03516751E-01
1 2 3.02903099E-01
2 1-1.55725013E-01-3.81037934E-01-1.55725013E-01-3.81037934E-01 3.11450027E-01
2 2 7.62075868E-01
3 1-2.69723635E-01-6.59977061E-01 2.69723635E-01 6.59977061E-01 0.00000000E+00
3 2 0.00000000E+00
4 1 5.27634310E-01-6.05171941E-01 5.27634310E-01-6.05171941E-01-1.05526862E+00
4 2 1.21034388E+00
5 1 9.13889433E-01-1.04818855E+00-9.13889433E-01 1.04818855E+00 0.00000000E+00
5 2 0.00000000E+00
6 1 7.72195430E-01-5.19424617E-01 7.72195430E-01-5.19424617E-01 7.72195430E-01
6 2-5.19424617E-01
$END
Output
--- RHF ORBITALS --- GENERATED AT 16:23:51 LT 4-DEC-2002
cycl H3_2plus S0
E(RHF)= -1.0977971729, E(NUC)= 0.9165618819, 7 ITERS
$VEC
1 1 2.03516751E-01 3.02903099E-01 2.03516751E-01 3.02903099E-01 2.03516751E-01
1 2 3.02903099E-01
2 1-1.55725013E-01-3.81037934E-01-1.55725013E-01-3.81037934E-01
3.11450027E-01
2 2 7.62075868E-01
3 1-2.69723635E-01-6.59977061E-01 2.69723635E-01 6.59977061E-01 0.00000000E+00
3 2 0.00000000E+00
4 1 5.27634310E-01-6.05171941E-01 5.27634310E-01-6.05171941E-01-1.05526862E+00
4 2 1.21034388E+00
5 1 9.13889433E-01-1.04818855E+00-9.13889433E-01 1.04818855E+00 0.00000000E+00
5 2 0.00000000E+00
6 1 7.72195430E-01-5.19424617E-01 7.72195430E-01-5.19424617E-01 7.72195430E-01
6 2-5.19424617E-01
$END
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
H 1.0 -1.6365507116 -0.9448629939 0.0000000000
H 1.0 1.6365507116 -0.9448629939 0.0000000000
H 1.0 0.0000000000 1.8897259877 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
H H H
1 H 0.0000000 1.7320508 * 1.7320508 *
2 H 1.7320508 * 0.0000000 1.7320508 *
3 H 1.7320508 * 1.7320508 * 0.0000000
ATOMIC BASIS SET
----------------
THE CONTRACTED PRIMITIVE FUNCTIONS HAVE BEEN UNNORMALIZED
THE CONTRACTED BASIS FUNCTIONS ARE NOW NORMALIZED TO UNITY
SHELL TYPE PRIM EXPONENT CONTRACTION COEFFICIENTS
H
5 S 1 18.731137 0.214935 ( 0.033495)
5 S 2 2.825394 0.364571 ( 0.234727)
5 S 3 0.640122 0.415051 ( 0.813757)
6 S 4 0.161278 0.181381 ( 1.000000)
TOTAL NUMBER OF SHELLS = 6
TOTAL NUMBER OF BASIS FUNCTIONS = 6
NUMBER OF ELECTRONS = 2
CHARGE OF MOLECULE = 1
STATE MULTIPLICITY = 3
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 2
NUMBER OF OCCUPIED ORBITALS (BETA ) = 0
TOTAL NUMBER OF ATOMS = 3
THE NUCLEAR REPULSION ENERGY IS 0.9165618819
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 0.9165618819
MAXIT = 30 NPUNCH= 2 MULT= 3
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
MEMORY REQUIRED FOR UHF/ROHF STEP= 8387 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 -0.829776548 -0.829776548 0.315494821 0.000000000
2 1 -0.789731924 0.040044623 0.392584143 0.000000000
3 2 -0.869064124 -0.079332200 0.258473239 0.000000000
4 3 -0.925100862 -0.056036738 0.203686704 0.000000000
5 4 -0.963581525 -0.038480663 0.132956890 0.000000000
6 0 -0.983334269 -0.019752744 0.412286245 0.000000000
7 1 -0.813432968 0.169901300 0.555303710 0.000000000
8 2 -0.958532473 -0.145099505 0.181969523 0.000000000
9 3 -0.955999166 0.002533307 0.164221026 0.000000000
10 4 -0.978868675 -0.022869509 0.155392028 0.000000000
11 5 -0.986403107 -0.007534432 0.109853820 0.000000000
12 6 -0.985072695 0.001330412 0.038074704 0.000000000
13 0 -0.983015226 0.002057469 0.028897417 0.000000000
14 1 -0.980470955 0.002544271 0.305942314 0.000000000
15 2 -0.834409721 0.146061233 0.643682302 0.000000000
16 3 -0.953775899 -0.119366178 0.218023544 0.000000000
17 4 -0.969017444 -0.015241545 0.171028068 0.000000000
18 0 -0.984277631 -0.015260187 0.608141095 0.000000000
19 1 -0.349106359 0.635171272 0.432862240 0.000000000
20 2 -0.765809089 -0.416702730 0.070858125 0.000000000
21 3 -0.786763112 -0.020954023 0.023119072 0.000000000
22 0 -0.787947135 -0.001184023 0.009115051 0.000000000
23 1 -0.788028017 -0.000080883 0.000151447 0.000000000
24 2 -0.788028107 -0.000000089 0.000042702 0.000000000
25 3 -0.788028111 -0.000000005 0.000012038 0.000000000
26 4 -0.788028111 0.000000000 0.000003393 0.000000000
27 5 -0.788028111 0.000000000 0.000000956 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -0.7880281115 AFTER 27 ITERATIONS
--------------------
SPIN SZ = 1.000
S-SQUARED = 2.000
--------------------
------------
EIGENVECTORS
------------
1 2
3 4
5
-0.7149
-0.7149 -0.6519 0.5882
0.5882
E' E'
A' E'
E'
1 H 1 S -0.295302 -0.295501 0.238147
0.307332 -0.970369
2 H 1 S -0.448346 -0.448648 0.279161
-0.387065 1.222118
3 H 2 S -0.108260 0.403490 0.238147
0.686698 0.751342
4 H 2 S -0.164368 0.612603 0.279161
-0.864853 -0.946267
5 H 3 S 0.403563 -0.107989 0.238147
-0.994030 0.219027
6 H 3 S 0.612713 -0.163955 0.279161
1.251918 -0.275851
MO1
MO1
MO2



MO3
MO3
MO4



6
0.6690
A'
1 H 1 S 0.762227
2 H 1 S -0.532561
3 H 2 S 0.762227
4 H 2 S -0.532561
5 H 3 S 0.762227
6 H 3 S -0.532561
...... END OF ROHF CALCULATION ......
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -1.9792972849
TWO ELECTRON ENERGY = 0.2747072915
NUCLEAR REPULSION ENERGY = 0.9165618819
------------------
TOTAL ENERGY = -0.7880281115
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.2747072915
NUCLEUS-ELECTRON POTENTIAL ENERGY = -3.3726046501
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 0.9165618819
------------------
TOTAL POTENTIAL ENERGY = -2.1813354766
TOTAL KINETIC ENERGY = 1.3933073651
VIRIAL RATIO (V/T) = 1.5655809559
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2
1.000000 1.000000
1 0.333109 0.333558
2 0.044770 0.621896
3 0.622121 0.044546
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H 0.666667 0.666667
2 H 0.666667 0.666667
3 H 0.666667 0.666667
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.30227 0.34279
2 H 1 S 0.36439 0.32387
3 H 2 S 0.30227 0.34279
4 H 2 S 0.36439 0.32387
5 H 3 S 0.30227 0.34279
6 H 3 S 0.36439 0.32387
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 0.9256836
2 -0.1295085 0.9256836
3 -0.1295085 -0.1295085 0.9256836
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H 0.666667 0.333333 0.666667 0.333333
2 H 0.666667 0.333333 0.666667 0.333333
3 H 0.666667 0.333333 0.666667 0.333333
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.732 0.222 1 3 1.732 0.222 2 3 1.732 0.222
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.889 0.444 0.444
2 H 0.889 0.444 0.444
3 H 0.889 0.444 0.444
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H 1.0 0.2600077
2 H 1.0 0.2600077
3 H 1.0 0.2600077