| Startseite |
Input
!
! Butadien
!
$CONTRL SCFTYP=RHF MULT=1 RUNTYP=OPTIMIZE COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
Butadien
CNH 2
C1
C2 1 1.40
C3 2 1.50 1 120.0
C4 3 1.40 2 120.0 1 180.0
H5 1 1.10 2 120.0 3 0.0
H6 1 1.10 2 120.0 3 180.0
H7 2 1.10 1 120.0 5 180.0
H8 3 1.10 2 120.0 1 0.0
H9 4 1.10 3 120.0 8 0.0
H10 4 1.10 3 120.0 8 180.0
$END
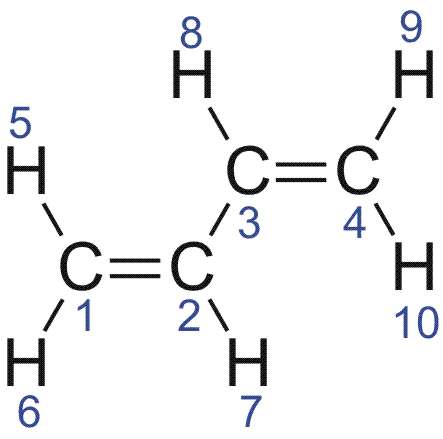
Die Z-Matrix entspricht folgender Struktur:
Output
TOTAL NUMBER OF BASIS FUNCTIONS = 26
NUMBER OF ELECTRONS = 30
CHARGE OF MOLECULE = 0
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 15
NUMBER OF OCCUPIED ORBITALS (BETA ) = 15
TOTAL NUMBER OF ATOMS = 10
NSERCH= 11 ENERGY= -154.5219567
-----------------------
GRADIENT (HARTREE/BOHR)
-----------------------
ATOM ZNUC DE/DX DE/DY DE/DZ
--------------------------------------------------------------
1 C1 6.0 -0.0000065 -0.0000149 0.0000000
2 C1 6.0 0.0000065 0.0000149 0.0000000
3 C2 6.0 -0.0000380 -0.0000557 0.0000000
4 C2 6.0 0.0000380 0.0000557 0.0000000
5 H5 1.0 0.0000601 -0.0000374 0.0000000
6 H5 1.0 -0.0000601 0.0000374 0.0000000
7 H6 1.0 0.0000055 0.0000068 0.0000000
8 H6 1.0 -0.0000055 -0.0000068 0.0000000
9 H7 1.0 -0.0000271 -0.0000087 0.0000000
10 H7 1.0 0.0000271 0.0000087 0.0000000
MAXIMUM GRADIENT = 0.0000601 RMS GRADIENT = 0.0000267
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
Butadien
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 -1.8322258550 0.1178444535 0.0000000000
C2 6.0 -0.6266676888 -0.4010225465 0.0000000000
H5 1.0 -1.9958802998 1.1828357691 0.0000000000
H6 1.0 -2.7176615957 -0.4953421525 0.0000000000
H7 1.0 -0.4957701083 -1.4734477683 0.0000000000
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 1.8322258550 -0.1178444535 0.0000000000
C1 6.0 -1.8322258550 0.1178444535 0.0000000000
C2 6.0 0.6266676888 0.4010225465 0.0000000000
C2 6.0 -0.6266676888 -0.4010225465 0.0000000000
H5 1.0 1.9958802998 -1.1828357691 0.0000000000
H5 1.0 -1.9958802998 1.1828357691 0.0000000000
H6 1.0 2.7176615957 0.4953421525 0.0000000000
H6 1.0 -2.7176615957 -0.4953421525 0.0000000000
H7 1.0 0.4957701083 1.4734477683 0.0000000000
H7 1.0 -0.4957701083 -1.4734477683 0.0000000000
THE CURRENT FULLY SUBSTITUTED Z-MATRIX IS
C1
C2 1 1.3124761
C2 2 1.4879939 1 124.0968271
C1 3 1.3124761 2 124.0968271 1 180.0000000 0
H5 1 1.0774921 2 122.0229909 3 0.0000000 0
H6 1 1.0770303 2 122.0095869 3 180.0000000 0
H7 2 1.0803842 1 120.2457937 5 180.0000000 0
H7 3 1.0803842 2 115.6573792 1 0.0000000 0
H6 4 1.0770303 3 122.0095869 8 0.0000000 0
H5 4 1.0774921 3 122.0229909 8 180.0000000 0
Nach der Geometrieoptimierung wird die Nummerierung der Z-Matrix geändert (bzw. das "Label" erzeugt von "Molekel" stimmt nicht mehr): C1-Typ: äußere C-Atome, C2-Typ: innere C-Atome.
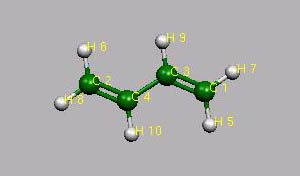
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
C1 C1 C2 C2
1 C1 0.0000000 3.6720234 1.3124761 * 2.4751459 *
2 C1 3.6720234 0.0000000 2.4751459 * 1.3124761 *
3 C2 1.3124761 * 2.4751459 * 0.0000000 1.4879939 *
4 C2 2.4751459 * 1.3124761 * 1.4879939 * 0.0000000
5 H5 1.0774921 * 4.0430392 2.0936452 * 2.7366019 *
6 H5 4.0430392 1.0774921 * 2.7366019 * 2.0936452 *
7 H6 1.0770303 * 4.5655208 2.0931201 * 3.4623703
8 H6 4.5655208 1.0770303 * 3.4623703 2.0931201 *
9 H7 2.0780580 * 2.6939238 * 1.0803842 * 2.1848354 *
10 H7 2.6939238 * 2.0780580 * 2.1848354 * 1.0803842 *
H5 H5 H6 H6
1 C1 1.0774921 * 4.0430392 1.0770303 * 4.5655208
2 C1 4.0430392 1.0774921 * 4.5655208 1.0770303 *
3 C2 2.0936452 * 2.7366019 * 2.0931201 * 3.4623703
4 C2 2.7366019 * 2.0936452 * 3.4623703 2.0931201 *
5 H5 0.0000000 4.6401029 1.8268140 * 4.7634152
6 H5 4.6401029 0.0000000 4.7634152 1.8268140 *
7 H6 1.8268140 * 4.7634152 0.0000000 5.5248705
8 H6 4.7634152 1.8268140 * 5.5248705 0.0000000
9 H7 3.0506020 2.5085408 * 2.4276516 * 3.7685909
10 H7 2.5085408 * 3.0506020 3.7685909 2.4276516 *
H7 H7
1 C1 2.0780580 * 2.6939238 *
2 C1 2.6939238 * 2.0780580 *
3 C2 1.0803842 * 2.1848354 *
4 C2 2.1848354 * 1.0803842 *
5 H5 3.0506020 2.5085408 *
6 H5 2.5085408 * 3.0506020
7 H6 2.4276516 * 3.7685909
8 H6 3.7685909 2.4276516 *
9 H7 0.0000000 3.1092355
10 H7 3.1092355 0.0000000
* ... LESS THAN 3.000
NUCLEAR ENERGY = 104.2502700377
ELECTRONIC ENERGY = -258.7722267128
TOTAL ENERGY = -154.5219566751
------------------
MOLECULAR ORBITALS
------------------
1 2 3 4 5
-11.1836 -11.1835 -11.1702 -11.1701 -1.0384
AG BU BU AG AG
1 C 1 S 0.032494 0.034312 0.702931 0.703021 -0.102115
2 C 1 S -0.002932 -0.002881 0.016719 0.016678 0.271921
3 C 1 X 0.002052 0.002061 -0.000939 -0.000898 -0.082023
4 C 1 Y -0.000966 -0.000788 0.000388 0.000409 0.028872
5 C 1 Z 0.000000 0.000000 0.000000 0.000000 0.000000
6 C 2 S 0.032494 -0.034312 -0.702931 0.703021 -0.102115
7 C 2 S -0.002932 0.002881 -0.016719 0.016678 0.271921
8 C 2 X -0.002052 0.002061 -0.000939 0.000898 0.082023
9 C 2 Y 0.000966 -0.000788 0.000388 -0.000409 -0.028872
10 C 2 Z 0.000000 0.000000 0.000000 0.000000 0.000000
11 C 3 S 0.703219 0.702748 -0.034179 -0.032346 -0.142827
12 C 3 S 0.013621 0.018842 -0.004678 -0.004292 0.384919
13 C 3 X 0.002386 -0.000982 -0.002066 -0.002191 0.008268
14 C 3 Y 0.000681 -0.001273 0.000967 0.000882 -0.052528
15 C 3 Z 0.000000 0.000000 0.000000 0.000000 0.000000
16 C 4 S 0.703219 -0.702748 0.034179 -0.032346 -0.142827
17 C 4 S 0.013621 -0.018842 0.004678 -0.004292 0.384919
18 C 4 X -0.002386 -0.000982 -0.002066 0.002191 -0.008268
19 C 4 Y -0.000681 -0.001273 0.000967 -0.000882 0.052528
20 C 4 Z 0.000000 0.000000 0.000000 0.000000 0.000000
21 H 5 S -0.000230 -0.000118 -0.003142 -0.003136 0.060138
22 H 6 S -0.000230 0.000118 0.003142 -0.003136 0.060138
23 H 7 S -0.000150 -0.000177 -0.003141 -0.003151 0.055826
24 H 8 S -0.000150 0.000177 0.003141 -0.003151 0.055826
25 H 9 S -0.003169 -0.003123 0.000200 0.000057 0.084944
26 H 10 S -0.003169 0.003123 -0.000200 0.000057 0.084944
6 7 8 9 10
-0.9564 -0.7733 -0.7135 -0.6003 -0.5953
BU AG BU BU AG
1 C 1 S -0.139796 -0.114407 0.053039 -0.017277 0.006245
2 C 1 S 0.384555 0.349796 -0.169558 0.078725 -0.030431
3 C 1 X -0.068193 0.125584 -0.100857 0.357576 0.036613
4 C 1 Y 0.034678 -0.021655 0.153286 0.045246 0.322407
5 C 1 Z 0.000000 0.000000 0.000000 0.000000 0.000000
6 C 2 S 0.139796 -0.114407 -0.053039 0.017277 0.006245
7 C 2 S -0.384555 0.349796 0.169558 -0.078725 -0.030431
8 C 2 X -0.068193 -0.125584 -0.100857 0.357576 -0.036613
9 C 2 Y 0.034678 0.021655 0.153286 0.045246 -0.322407
10 C 2 Z 0.000000 0.000000 0.000000 0.000000 0.000000
11 C 3 S -0.095581 0.082243 -0.090930 0.039574 -0.006426
12 C 3 S 0.261119 -0.258032 0.285900 -0.115096 0.017547
13 C 3 X 0.153606 0.194445 0.004880 -0.171925 0.185810
14 C 3 Y -0.018128 -0.002374 0.217294 0.202292 0.217338
15 C 3 Z 0.000000 0.000000 0.000000 0.000000 0.000000
16 C 4 S 0.095581 0.082243 0.090930 -0.039574 -0.006426
17 C 4 S -0.261119 -0.258032 -0.285900 0.115096 0.017547
18 C 4 X 0.153606 -0.194445 0.004880 -0.171925 -0.185810
19 C 4 Y -0.018128 0.002374 0.217294 0.202292 -0.217338
20 C 4 Z 0.000000 0.000000 0.000000 0.000000 0.000000
21 H 5 S 0.098838 0.156934 -0.181943 0.039591 -0.254068
22 H 6 S -0.098838 0.156934 0.181943 -0.039591 -0.254068
23 H 7 S 0.103314 0.187098 -0.068840 0.282530 0.154299
24 H 8 S -0.103314 0.187098 0.068840 -0.282530 0.154299
25 H 9 S 0.062911 -0.109551 0.259927 0.114218 0.152701
26 H 10 S -0.062911 -0.109551 -0.259927 -0.114218 0.152701
11 12 13 14 15
-0.5082 -0.5027 -0.4368 -0.4021 -0.2804
AG BU AG AU BG
1 C 1 S 0.025969 -0.006322 -0.000767 0.000000 0.000000
2 C 1 S -0.069452 0.011181 -0.005153 0.000000 0.000000
3 C 1 X 0.351128 0.015753 -0.128163 0.000000 0.000000
4 C 1 Y -0.093473 0.362085 -0.201874 0.000000 0.000000
5 C 1 Z 0.000000 0.000000 0.000000 0.372065 0.520906
6 C 2 S 0.025969 0.006322 -0.000767 0.000000 0.000000
7 C 2 S -0.069452 -0.011181 -0.005153 0.000000 0.000000
8 C 2 X -0.351128 0.015753 0.128163 0.000000 0.000000
9 C 2 Y 0.093473 0.362085 0.201874 0.000000 0.000000
10 C 2 Z 0.000000 0.000000 0.000000 0.372065 -0.520906
11 C 3 S 0.002570 0.019464 0.019840 0.000000 0.000000
12 C 3 S 0.015681 -0.083731 -0.085015 0.000000 0.000000
13 C 3 X -0.327931 0.125414 0.232337 0.000000 0.000000
14 C 3 Y 0.190767 -0.165595 0.327864 0.000000 0.000000
15 C 3 Z 0.000000 0.000000 0.000000 0.474426 0.403905
16 C 4 S 0.002570 -0.019464 0.019840 0.000000 0.000000
17 C 4 S 0.015681 0.083731 -0.085015 0.000000 0.000000
18 C 4 X 0.327931 0.125414 -0.232337 0.000000 0.000000
19 C 4 Y -0.190767 -0.165595 -0.327864 0.000000 0.000000
20 C 4 Z 0.000000 0.000000 0.000000 0.474426 -0.403905
21 H 5 S 0.103458 -0.321185 0.214952 0.000000 0.000000
22 H 6 S 0.103458 0.321185 0.214952 0.000000 0.000000
23 H 7 S 0.179638 0.218111 -0.243452 0.000000 0.000000
24 H 8 S 0.179638 -0.218111 -0.243452 0.000000 0.000000
25 H 9 S 0.220725 -0.202204 0.290269 0.000000 0.000000
26 H 10 S 0.220725 0.202204 0.290269 0.000000 0.000000
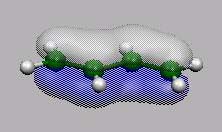
MO14
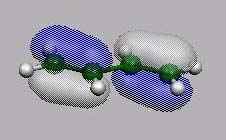
MO15
16 17 18 19 20
0.2491 0.4114 0.5737 0.6668 0.6784
AU BG BU AG BU
1 C 1 S 0.000000 0.000000 -0.018023 -0.006333 0.034258
2 C 1 S 0.000000 0.000000 0.118789 0.033248 -0.193690
3 C 1 X 0.000000 0.000000 0.088757 -0.388895 -0.004297
4 C 1 Y 0.000000 0.000000 0.455809 -0.373967 -0.075585
5 C 1 Z 0.627770 -0.512540 0.000000 0.000000 0.000000
6 C 2 S 0.000000 0.000000 0.018023 -0.006333 -0.034258
7 C 2 S 0.000000 0.000000 -0.118789 0.033248 0.193690
8 C 2 X 0.000000 0.000000 0.088757 0.388895 -0.004297
9 C 2 Y 0.000000 0.000000 0.455809 0.373967 -0.075585
10 C 2 Z 0.627770 0.512540 0.000000 0.000000 0.000000
11 C 3 S 0.000000 0.000000 0.030212 0.073693 -0.155429
12 C 3 S 0.000000 0.000000 -0.192900 -0.537477 1.034016
13 C 3 X 0.000000 0.000000 0.249634 -0.021363 -0.488203
14 C 3 Y 0.000000 0.000000 0.553996 -0.383528 0.117489
15 C 3 Z -0.476023 0.698337 0.000000 0.000000 0.000000
16 C 4 S 0.000000 0.000000 -0.030212 0.073693 0.155429
17 C 4 S 0.000000 0.000000 0.192900 -0.537477 -1.034016
18 C 4 X 0.000000 0.000000 0.249634 0.021363 -0.488203
19 C 4 Y 0.000000 0.000000 0.553996 0.383528 0.117489
20 C 4 Z -0.476023 -0.698337 0.000000 0.000000 0.000000
21 H 5 S 0.000000 0.000000 0.407836 -0.367700 0.015035
22 H 6 S 0.000000 0.000000 -0.407836 -0.367700 -0.015035
23 H 7 S 0.000000 0.000000 -0.447795 0.496633 0.175345
24 H 8 S 0.000000 0.000000 0.447795 0.496633 -0.175345
25 H 9 S 0.000000 0.000000 -0.455306 0.627890 -0.707703
26 H 10 S 0.000000 0.000000 0.455306 0.627890 0.707703
MO16
MO17
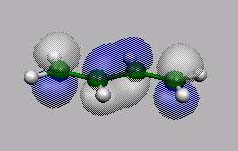
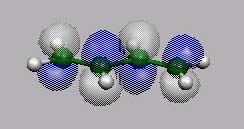
MO14, MO15,
MO16 und MO17 sind die bekannten 4
p-MO's von Butadien.
21 22 23 24 25
0.6948 0.7073 0.8799 0.9254 1.0065
BU AG AG BU BU
1 C 1 S -0.135716 -0.129265 -0.032115 0.053287 0.040220
2 C 1 S 0.965057 0.914127 0.244372 -0.443538 -0.365532
3 C 1 X 0.280469 0.343980 0.037725 0.821347 0.279548
4 C 1 Y -0.169856 -0.228037 0.704858 0.178072 -0.676382
5 C 1 Z 0.000000 0.000000 0.000000 0.000000 0.000000
6 C 2 S 0.135716 -0.129265 -0.032115 -0.053287 -0.040220
7 C 2 S -0.965057 0.914127 0.244372 0.443538 0.365532
8 C 2 X 0.280469 -0.343980 -0.037725 0.821347 0.279548
9 C 2 Y -0.169856 0.228037 -0.704858 0.178072 -0.676382
10 C 2 Z 0.000000 0.000000 0.000000 0.000000 0.000000
11 C 3 S 0.006099 0.021262 0.075619 -0.087587 -0.021112
12 C 3 S -0.014742 -0.116549 -0.601351 0.708619 0.227928
13 C 3 X -0.242234 -0.107483 -0.222393 0.269640 0.822101
14 C 3 Y 0.065606 0.030072 -0.444113 -0.550374 0.380512
15 C 3 Z 0.000000 0.000000 0.000000 0.000000 0.000000
16 C 4 S -0.006099 0.021262 0.075619 0.087587 0.021112
17 C 4 S 0.014742 -0.116549 -0.601351 -0.708619 -0.227928
18 C 4 X -0.242234 0.107483 0.222393 0.269640 0.822101
19 C 4 Y 0.065606 -0.030072 0.444113 -0.550374 0.380512
20 C 4 Z 0.000000 0.000000 0.000000 0.000000 0.000000
21 H 5 S -0.664845 -0.702054 0.421776 0.211944 -0.394407
22 H 6 S 0.664845 -0.702054 0.421776 -0.211944 0.394407
23 H 7 S -0.587854 -0.580282 -0.453781 -0.422298 0.241434
24 H 8 S 0.587854 -0.580282 -0.453781 0.422298 -0.241434
25 H 9 S -0.090218 0.077033 0.542887 0.166653 -0.282972
26 H 10 S 0.090218 0.077033 0.542887 -0.166653 0.282972
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -414.5149059052
TWO ELECTRON ENERGY = 155.7426791924
NUCLEAR REPULSION ENERGY = 104.2502700377
------------------
TOTAL ENERGY = -154.5219566751
ELECTRON-ELECTRON POTENTIAL ENERGY = 155.7426791924
NUCLEUS-ELECTRON POTENTIAL ENERGY = -569.1307423800
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 104.2502700377
------------------
TOTAL POTENTIAL ENERGY = -309.1377931499
TOTAL KINETIC ENERGY = 154.6158364748
VIRIAL RATIO (V/T) = 1.9993928190
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -103.0295565420
BARE H ENERGY= -414.5149059052
ELECTRONIC ENERGY = -258.7722312236
KINETIC ENERGY= 154.6158364748
N-N REPULSION= 104.2502700377
TOTAL ENERGY= -154.5219611860
SIGMA PART(1+2)= -237.8389681317
(K,V1,2)= 149.5619021006 -523.5753857646 136.1745155323
PI PART(1+2)= -20.9332630919
(K,V1,2)= 5.0539343742 -45.5553566153 19.5681591492
SIGMA SKELETON, ERROR= -133.5886980940 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.001561 0.001858 0.998809 0.999045 0.321932
2 0.001561 0.001858 0.998809 0.999045 0.321932
3 0.998789 0.998435 0.001820 0.001585 0.564792
4 0.998789 0.998435 0.001820 0.001585 0.564792
5 -0.000004 -0.000001 -0.000315 -0.000314 0.029624
6 -0.000004 -0.000001 -0.000315 -0.000314 0.029624
7 -0.000002 -0.000003 -0.000315 -0.000316 0.024825
8 -0.000002 -0.000003 -0.000315 -0.000316 0.024825
9 -0.000344 -0.000289 0.000001 0.000000 0.058828
10 -0.000344 -0.000289 0.000001 0.000000 0.058828
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.502212 0.374271 0.210218 0.448094 0.373670
2 0.502212 0.374271 0.210218 0.448094 0.373670
3 0.342114 0.318029 0.378699 0.248881 0.284701
4 0.342114 0.318029 0.378699 0.248881 0.284701
5 0.062305 0.105684 0.132262 0.005471 0.198037
6 0.062305 0.105684 0.132262 0.005471 0.198037
7 0.069464 0.152805 0.017960 0.253739 0.072747
8 0.069464 0.152805 0.017960 0.253739 0.072747
9 0.023905 0.049212 0.260860 0.043816 0.070844
10 0.023905 0.049212 0.260860 0.043816 0.070844
11 12 13 14 15
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.363858 0.381704 0.147179 0.372912 0.637311
2 0.363858 0.381704 0.147179 0.372912 0.637311
3 0.383761 0.129981 0.414337 0.627088 0.362689
4 0.383761 0.129981 0.414337 0.627088 0.362689
5 0.031551 0.265802 0.106840 0.000000 0.000000
6 0.031551 0.265802 0.106840 0.000000 0.000000
7 0.089677 0.119353 0.135056 0.000000 0.000000
8 0.089677 0.119353 0.135056 0.000000 0.000000
9 0.131154 0.103160 0.196588 0.000000 0.000000
10 0.131154 0.103160 0.196588 0.000000 0.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 C 1 S 1.99582 1.99014
2 C 1 S 1.13893 1.01465
3 C 1 X 0.98949 1.03540
4 C 1 Y 1.00017 1.02777
5 C 1 Z 1.01022 1.01087
6 C 2 S 1.99582 1.99014
7 C 2 S 1.13893 1.01465
8 C 2 X 0.98949 1.03540
9 C 2 Y 1.00017 1.02777
10 C 2 Z 1.01022 1.01087
11 C 3 S 1.99580 1.98985
12 C 3 S 1.12786 1.01699
13 C 3 X 0.96085 1.01305
14 C 3 Y 0.98143 1.01203
15 C 3 Z 0.98978 0.98913
16 C 4 S 1.99580 1.98985
17 C 4 S 1.12786 1.01699
18 C 4 X 0.96085 1.01305
19 C 4 Y 0.98143 1.01203
20 C 4 Z 0.98978 0.98913
21 H 5 S 0.93694 0.96638
22 H 6 S 0.93694 0.96638
23 H 7 S 0.93499 0.96498
24 H 8 S 0.93499 0.96498
25 H 9 S 0.93774 0.96875
26 H 10 S 0.93774 0.96875
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 4.7962777
2 0.0009095 4.7962777
3 0.6076333 -0.0278569 4.7543881
4 -0.0278569 0.6076333 0.4076276 4.7543881
5 0.3938541 0.0000593 -0.0257096 -0.0049506 0.5946446
6 0.0000593 0.3938541 -0.0049506 -0.0257096 0.0000036
7 0.3951158 -0.0000487 -0.0249387 0.0018140 -0.0240535
8 -0.0000487 0.3951158 0.0018140 -0.0249387 -0.0000054
9 -0.0279619 -0.0033483 0.3929225 -0.0252285 0.0024841
10 -0.0033483 -0.0279619 -0.0252285 0.3929225 0.0006137
6 7 8 9 10
6 0.5946446
7 -0.0000054 0.5925951
8 -0.0240535 0.0000025 0.5925951
9 0.0006137 -0.0054921 0.0000002 0.6019641
10 0.0024841 0.0000002 -0.0054921 0.0017816 0.6019641
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 C1 6.134634 -0.134634 6.078832 -0.078832
2 C1 6.134634 -0.134634 6.078832 -0.078832
3 C2 6.055701 -0.055701 6.021056 -0.021056
4 C2 6.055701 -0.055701 6.021056 -0.021056
5 H5 0.936940 0.063060 0.966384 0.033616
6 H5 0.936940 0.063060 0.966384 0.033616
7 H6 0.934989 0.065011 0.964976 0.035024
8 H6 0.934989 0.065011 0.964976 0.035024
9 H7 0.937735 0.062265 0.968752 0.031248
10 H7 0.937735 0.062265 0.968752 0.031248
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 3.672 0.072 1 3 1.312 1.945 1 5 1.077 0.977
1 7 1.077 0.978 2 4 1.312 1.945 2 6 1.077 0.977
2 8 1.077 0.978 3 4 1.488 1.042 3 9 1.080 0.970
4 10 1.080 0.970
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 C1 3.977 3.977 0.000
2 C1 3.977 3.977 0.000
3 C2 3.979 3.979 0.000
4 C2 3.979 3.979 0.000
5 H5 0.996 0.996 0.000
6 H5 0.996 0.996 0.000
7 H6 0.996 0.996 0.000
8 H6 0.996 0.996 0.000
9 H7 0.996 0.996 0.000
10 H7 0.996 0.996 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 0.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 0.000000 0.000000
...... END OF PROPERTY EVALUATION ......
| Seitenanfang |