| Startseite |
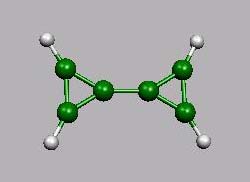
Das neutrale Molekül dient als einfaches Beispiel für ein System mit einer zentralen Doppelbindung (als Modell für die Drehung um eine Doppelbindung). Im Dikation wird diese Bindung zur formalen Einfachbindung.
Input
!
! Bicyclopropen
!
$CONTRL SCFTYP=RHF MULT=1 RUNTYP=OPTIMIZE COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
Bicyclopropen
CNH 2
C1
C2 1 1.40
C3 2 1.40 1 150.00
C4 2 1.40 1 150.00 3 180.00 0
C5 1 1.40 2 150.00 3 180.00 0
C6 1 1.40 2 150.00 3 0.00 0
H7 3 1.10 2 150.00 4 180.00 0
H8 4 1.10 2 150.00 3 180.00 0
H9 5 1.10 1 150.00 6 180.00 0
H10 6 1.10 1 150.00 5 180.00 0
$END
Output
TOTAL NUMBER OF SHELLS = 16
TOTAL NUMBER OF BASIS FUNCTIONS = 34
NUMBER OF ELECTRONS = 40
CHARGE OF MOLECULE = 0
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 20
NUMBER OF OCCUPIED ORBITALS (BETA ) = 20
TOTAL NUMBER OF ATOMS = 10
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 174.4674443816
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=T
DENSITY CONV= 1.00E-05
SOSCF WILL OPTIMIZE 280 ORBITAL ROTATIONS, SOGTOL= 0.250
MEMORY REQUIRED FOR RHF STEP= 17185 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE ORB. GRAD
1 0 0 -228.334309196 -228.334309196 0.259895534 0.000000000
---------------START SECOND ORDER SCF---------------
2 1 0 -228.590908047 -0.256598851 0.055217693 0.025245442
3 2 0 -228.598964961 -0.008056913 0.025482038 0.006629551
4 3 0 -228.599824308 -0.000859348 0.002160616 0.001403213
5 4 0 -228.599840702 -0.000016394 0.000214686 0.000230621
6 5 0 -228.599841068 -0.000000366 0.000088109 0.000072024
7 6 0 -228.599841097 -0.000000028 0.000021203 0.000011102
8 7 0 -228.599841098 -0.000000001 0.000003105 0.000002880
9 8 0 -228.599841098 0.000000000 0.000000516 0.000000327
-----------------
DENSITY CONVERGED
-----------------
NSERCH= 11 ENERGY= -228.6666888
-----------------------
GRADIENT (HARTREE/BOHR)
-----------------------
ATOM ZNUC DE/DX DE/DY DE/DZ
--------------------------------------------------------------
1 C1 6.0 0.0000000 0.0000000 -0.0000055
2 C1 6.0 0.0000000 0.0000000 0.0000055
3 C3 6.0 -0.0000040 0.0000000 0.0000029
4 C3 6.0 0.0000040 0.0000000 -0.0000029
5 C3 6.0 -0.0000040 0.0000000 -0.0000029
6 C3 6.0 0.0000040 0.0000000 0.0000029
7 H7 1.0 0.0000119 0.0000000 -0.0000036
8 H7 1.0 -0.0000119 0.0000000 0.0000036
9 H7 1.0 0.0000119 0.0000000 0.0000036
10 H7 1.0 -0.0000119 0.0000000 -0.0000036
MAXIMUM GRADIENT = 0.0000119 RMS GRADIENT = 0.0000051
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
Bicyclopropylium
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000 -0.6408513870
C3 6.0 0.6479786518 0.0000000000 1.9493238108
H7 1.0 1.5608971144 0.0000000000 2.5134576856
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000 0.6408513870
C1 6.0 0.0000000000 0.0000000000 -0.6408513870
C3 6.0 -0.6479786518 0.0000000000 1.9493238108
C3 6.0 0.6479786518 0.0000000000 -1.9493238108
C3 6.0 -0.6479786518 0.0000000000 -1.9493238108
C3 6.0 0.6479786518 0.0000000000 1.9493238108
H7 1.0 -1.5608971144 0.0000000000 2.5134576856
H7 1.0 1.5608971144 0.0000000000 -2.5134576856
H7 1.0 -1.5608971144 0.0000000000 -2.5134576856
H7 1.0 1.5608971144 0.0000000000 2.5134576856
THE CURRENT FULLY SUBSTITUTED Z-MATRIX IS
C1
C1 1 1.2817028
C3 2 1.4601289 1 153.6545742
C3 2 1.4601289 1 153.6545742 3 180.0000000 0
C3 1 1.4601289 2 153.6545742 3 180.0000000 0
C3 1 1.4601289 2 153.6545742 3 0.0000000 0
H7 3 1.0731576 2 148.0592322 4 180.0000000 0
H7 4 1.0731576 2 148.0592322 3 180.0000000 0
H7 5 1.0731576 1 148.0592322 6 180.0000000 0
H7 6 1.0731576 1 148.0592322 5 180.0000000 0
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
C1 C1 C3 C3
1 C1 0.0000000 1.2817028 * 1.4601289 * 2.6699970 *
2 C1 1.2817028 * 0.0000000 2.6699970 * 1.4601289 *
3 C3 1.4601289 * 2.6699970 * 0.0000000 4.1084010
4 C3 2.6699970 * 1.4601289 * 4.1084010 0.0000000
5 C3 2.6699970 * 1.4601289 * 3.8986476 1.2959573 *
6 C3 1.4601289 * 2.6699970 * 1.2959573 * 3.8986476
7 H7 2.4378380 * 3.5193843 1.0731576 * 4.9795131
8 H7 3.5193843 2.4378380 * 4.9795131 1.0731576 *
9 H7 3.5193843 2.4378380 * 4.5551991 2.2797761 *
10 H7 2.4378380 * 3.5193843 2.2797761 * 4.5551991
C3 C3 H7 H7
1 C1 2.6699970 * 1.4601289 * 2.4378380 * 3.5193843
2 C1 1.4601289 * 2.6699970 * 3.5193843 2.4378380 *
3 C3 3.8986476 1.2959573 * 1.0731576 * 4.9795131
4 C3 1.2959573 * 3.8986476 4.9795131 1.0731576 *
5 C3 0.0000000 4.1084010 4.5551991 2.2797761 *
6 C3 4.1084010 0.0000000 2.2797761 * 4.5551991
7 H7 4.5551991 2.2797761 * 0.0000000 5.9173877
8 H7 2.2797761 * 4.5551991 5.9173877 0.0000000
9 H7 1.0731576 * 4.9795131 5.0269154 3.1217942
10 H7 4.9795131 1.0731576 * 3.1217942 5.0269154
H7 H7
1 C1 3.5193843 2.4378380 *
2 C1 2.4378380 * 3.5193843
3 C3 4.5551991 2.2797761 *
4 C3 2.2797761 * 4.5551991
5 C3 1.0731576 * 4.9795131
6 C3 4.9795131 1.0731576 *
7 H7 5.0269154 3.1217942
8 H7 3.1217942 5.0269154
9 H7 0.0000000 5.9173877
10 H7 5.9173877 0.0000000
* ... LESS THAN 3.000
NUCLEAR ENERGY = 176.3284658827
ELECTRONIC ENERGY = -404.9951546739
TOTAL ENERGY = -228.6666887912
------------------
MOLECULAR ORBITALS
------------------
1 2 3 4 5
-11.1946 -11.1946 -11.1930 -11.1930 -11.1646
AG AU BG BU AG
1 C 1 S 0.005360 0.005523 0.000000 0.000000 0.703822
2 C 1 S -0.003518 -0.003342 0.000000 0.000000 0.012560
3 C 1 X 0.000000 0.000000 0.002085 0.001549 0.000000
4 C 1 Y 0.000000 0.000000 0.000000 0.000000 0.000000
5 C 1 Z -0.002051 -0.002124 0.000000 0.000000 0.002327
6 C 2 S 0.005360 -0.005523 0.000000 0.000000 0.703822
7 C 2 S -0.003518 0.003342 0.000000 0.000000 0.012560
8 C 2 X 0.000000 0.000000 -0.002085 0.001549 0.000000
9 C 2 Y 0.000000 0.000000 0.000000 0.000000 0.000000
10 C 2 Z 0.002051 -0.002124 0.000000 0.000000 -0.002327
11 C 3 S 0.497641 0.497641 0.497683 0.497694 -0.003547
12 C 3 S 0.008898 0.008888 0.013889 0.013755 -0.002277
13 C 3 X 0.000105 0.000105 0.003113 0.003077 -0.000243
14 C 3 Y 0.000000 0.000000 0.000000 0.000000 0.000000
15 C 3 Z -0.000968 -0.000960 -0.000217 -0.000114 0.001654
16 C 4 S 0.497641 -0.497641 0.497683 -0.497694 -0.003547
17 C 4 S 0.008898 -0.008888 0.013889 -0.013755 -0.002277
18 C 4 X -0.000105 0.000105 -0.003113 0.003077 0.000243
19 C 4 Y 0.000000 0.000000 0.000000 0.000000 0.000000
20 C 4 Z 0.000968 -0.000960 0.000217 -0.000114 -0.001654
21 C 5 S 0.497641 -0.497641 -0.497683 0.497694 -0.003547
22 C 5 S 0.008898 -0.008888 -0.013889 0.013755 -0.002277
23 C 5 X 0.000105 -0.000105 -0.003113 0.003077 -0.000243
24 C 5 Y 0.000000 0.000000 0.000000 0.000000 0.000000
25 C 5 Z 0.000968 -0.000960 -0.000217 0.000114 -0.001654
26 C 6 S 0.497641 0.497641 -0.497683 -0.497694 -0.003547
27 C 6 S 0.008898 0.008888 -0.013889 -0.013755 -0.002277
28 C 6 X -0.000105 -0.000105 0.003113 0.003077 0.000243
29 C 6 Y 0.000000 0.000000 0.000000 0.000000 0.000000
30 C 6 Z -0.000968 -0.000960 0.000217 0.000114 0.001654
31 H 7 S -0.001765 -0.001763 -0.001821 -0.001824 0.000004
32 H 8 S -0.001765 0.001763 -0.001821 0.001824 0.000004
33 H 9 S -0.001765 0.001763 0.001821 -0.001824 0.000004
34 H 10 S -0.001765 -0.001763 0.001821 0.001824 0.000004
6 7 8 9 10
-11.1632 -1.1279 -1.1035 -0.8530 -0.7329
AU AG AU AG BU
1 C 1 S 0.703751 -0.109223 -0.073181 -0.138528 0.000000
2 C 1 S 0.019302 0.263558 0.146478 0.392300 0.000000
3 C 1 X 0.000000 0.000000 0.000000 0.000000 -0.134766
4 C 1 Y 0.000000 0.000000 0.000000 0.000000 0.000000
5 C 1 Z -0.001406 0.043212 0.143943 -0.255108 0.000000
6 C 2 S -0.703751 -0.109223 0.073181 -0.138528 0.000000
7 C 2 S -0.019302 0.263558 -0.146478 0.392300 0.000000
8 C 2 X 0.000000 0.000000 0.000000 0.000000 -0.134766
9 C 2 Y 0.000000 0.000000 0.000000 0.000000 0.000000
10 C 2 Z -0.001406 -0.043212 0.143943 0.255108 0.000000
11 C 3 S -0.003633 -0.101385 -0.111689 0.061711 -0.090384
12 C 3 S -0.002330 0.246255 0.271134 -0.169553 0.290754
13 C 3 X -0.000218 0.106545 0.117482 -0.062613 -0.151953
14 C 3 Y 0.000000 0.000000 0.000000 0.000000 0.000000
15 C 3 Z 0.001697 -0.056643 -0.049713 -0.080171 0.009933
16 C 4 S 0.003633 -0.101385 0.111689 0.061711 0.090384
17 C 4 S 0.002330 0.246255 -0.271134 -0.169553 -0.290754
18 C 4 X -0.000218 -0.106545 0.117482 0.062613 -0.151953
19 C 4 Y 0.000000 0.000000 0.000000 0.000000 0.000000
20 C 4 Z 0.001697 0.056643 -0.049713 0.080171 0.009933
21 C 5 S 0.003633 -0.101385 0.111689 0.061711 -0.090384
22 C 5 S 0.002330 0.246255 -0.271134 -0.169553 0.290754
23 C 5 X 0.000218 0.106545 -0.117482 -0.062613 -0.151953
24 C 5 Y 0.000000 0.000000 0.000000 0.000000 0.000000
25 C 5 Z 0.001697 0.056643 -0.049713 0.080171 -0.009933
26 C 6 S -0.003633 -0.101385 -0.111689 0.061711 0.090384
27 C 6 S -0.002330 0.246255 0.271134 -0.169553 -0.290754
28 C 6 X 0.000218 -0.106545 -0.117482 0.062613 -0.151953
29 C 6 Y 0.000000 0.000000 0.000000 0.000000 0.000000
30 C 6 Z 0.001697 -0.056643 -0.049713 -0.080171 -0.009933
31 H 7 S 0.000025 0.042381 0.048863 -0.063067 0.205345
32 H 8 S -0.000025 0.042381 -0.048863 -0.063067 -0.205345
33 H 9 S -0.000025 0.042381 -0.048863 -0.063067 0.205345
34 H 10 S 0.000025 0.042381 0.048863 -0.063067 -0.205345
11 12 13 14 15
-0.7230 -0.6891 -0.6599 -0.5077 -0.4824
BG AU AG AU BU
1 C 1 S 0.000000 0.062958 -0.007980 0.066321 0.000000
2 C 1 S 0.000000 -0.176584 0.026970 -0.271234 0.000000
3 C 1 X -0.092972 0.000000 0.000000 0.000000 0.000000
4 C 1 Y 0.000000 0.000000 0.000000 0.000000 0.403260
5 C 1 Z 0.000000 -0.123217 -0.211677 -0.111483 0.000000
6 C 2 S 0.000000 -0.062958 -0.007980 -0.066321 0.000000
7 C 2 S 0.000000 0.176584 0.026970 0.271234 0.000000
8 C 2 X 0.092972 0.000000 0.000000 0.000000 0.000000
9 C 2 Y 0.000000 0.000000 0.000000 0.000000 0.403260
10 C 2 Z 0.000000 -0.123217 0.211677 -0.111483 0.000000
11 C 3 S -0.090846 -0.043204 -0.023668 -0.016570 0.000000
12 C 3 S 0.292187 0.167436 0.114522 0.044072 0.000000
13 C 3 X -0.158625 -0.127164 -0.204535 0.319477 0.000000
14 C 3 Y 0.000000 0.000000 0.000000 0.000000 0.283018
15 C 3 Z 0.024346 0.210706 0.164541 0.192742 0.000000
16 C 4 S -0.090846 0.043204 -0.023668 0.016570 0.000000
17 C 4 S 0.292187 -0.167436 0.114522 -0.044072 0.000000
18 C 4 X 0.158625 -0.127164 0.204535 0.319477 0.000000
19 C 4 Y 0.000000 0.000000 0.000000 0.000000 0.283018
20 C 4 Z -0.024346 0.210706 -0.164541 0.192742 0.000000
21 C 5 S 0.090846 0.043204 -0.023668 0.016570 0.000000
22 C 5 S -0.292187 -0.167436 0.114522 -0.044072 0.000000
23 C 5 X 0.158625 0.127164 -0.204535 -0.319477 0.000000
24 C 5 Y 0.000000 0.000000 0.000000 0.000000 0.283018
25 C 5 Z 0.024346 0.210706 -0.164541 0.192742 0.000000
26 C 6 S 0.090846 -0.043204 -0.023668 -0.016570 0.000000
27 C 6 S -0.292187 0.167436 0.114522 0.044072 0.000000
28 C 6 X -0.158625 0.127164 0.204535 -0.319477 0.000000
29 C 6 Y 0.000000 0.000000 0.000000 0.000000 0.283018
30 C 6 Z -0.024346 0.210706 0.164541 0.192742 0.000000
31 H 7 S 0.218062 0.205216 0.215330 -0.132878 0.000000
32 H 8 S 0.218062 -0.205216 0.215330 0.132878 0.000000
33 H 9 S -0.218062 -0.205216 0.215330 0.132878 0.000000
34 H 10 S -0.218062 0.205216 0.215330 -0.132878 0.000000
16 17 18 19 20
-0.4566 -0.4520 -0.3995 -0.3190 -0.1759
BU AG BG BG BU
1 C 1 S 0.000000 0.000738 0.000000 0.000000 0.000000
2 C 1 S 0.000000 0.055779 0.000000 0.000000 0.000000
3 C 1 X 0.425261 0.000000 0.000000 0.398021 0.000000
4 C 1 Y 0.000000 0.000000 0.194400 0.000000 0.505334
5 C 1 Z 0.000000 0.315715 0.000000 0.000000 0.000000
6 C 2 S 0.000000 0.000738 0.000000 0.000000 0.000000
7 C 2 S 0.000000 0.055779 0.000000 0.000000 0.000000
8 C 2 X 0.425261 0.000000 0.000000 -0.398021 0.000000
9 C 2 Y 0.000000 0.000000 -0.194400 0.000000 0.505334
10 C 2 Z 0.000000 -0.315715 0.000000 0.000000 0.000000
11 C 3 S 0.011032 0.009586 0.000000 0.035807 0.000000
12 C 3 S -0.076594 -0.035865 0.000000 -0.194526 0.000000
13 C 3 X -0.060522 -0.274705 0.000000 -0.033600 0.000000
14 C 3 Y 0.000000 0.000000 0.407302 0.000000 -0.360278
15 C 3 Z 0.243565 -0.244878 0.000000 0.329515 0.000000
16 C 4 S -0.011032 0.009586 0.000000 0.035807 0.000000
17 C 4 S 0.076594 -0.035865 0.000000 -0.194526 0.000000
18 C 4 X -0.060522 0.274705 0.000000 0.033600 0.000000
19 C 4 Y 0.000000 0.000000 -0.407302 0.000000 -0.360278
20 C 4 Z 0.243565 0.244878 0.000000 -0.329515 0.000000
21 C 5 S 0.011032 0.009586 0.000000 -0.035807 0.000000
22 C 5 S -0.076594 -0.035865 0.000000 0.194526 0.000000
23 C 5 X -0.060522 -0.274705 0.000000 0.033600 0.000000
24 C 5 Y 0.000000 0.000000 -0.407302 0.000000 -0.360278
25 C 5 Z -0.243565 0.244878 0.000000 0.329515 0.000000
26 C 6 S -0.011032 0.009586 0.000000 -0.035807 0.000000
27 C 6 S 0.076594 -0.035865 0.000000 0.194526 0.000000
28 C 6 X -0.060522 0.274705 0.000000 -0.033600 0.000000
29 C 6 Y 0.000000 0.000000 0.407302 0.000000 -0.360278
30 C 6 Z -0.243565 -0.244878 0.000000 -0.329515 0.000000
31 H 7 S 0.164162 0.101247 0.000000 0.171681 0.000000
32 H 8 S -0.164162 0.101247 0.000000 0.171681 0.000000
33 H 9 S 0.164162 0.101247 0.000000 -0.171681 0.000000
34 H 10 S -0.164162 0.101247 0.000000 -0.171681 0.000000
21 22 23 24 25
0.3127 0.3137 0.4214 0.4245 0.5712
AG AU BU BG AU
1 C 1 S 0.000000 0.000000 0.000000 0.000000 -0.157365
2 C 1 S 0.000000 0.000000 0.000000 0.000000 0.986405
3 C 1 X 0.000000 0.000000 0.519771 0.000000 0.000000
4 C 1 Y 0.000000 0.000000 0.000000 0.829983 0.000000
5 C 1 Z 0.000000 0.000000 0.000000 0.000000 0.116073
6 C 2 S 0.000000 0.000000 0.000000 0.000000 0.157365
7 C 2 S 0.000000 0.000000 0.000000 0.000000 -0.986405
8 C 2 X 0.000000 0.000000 0.519771 0.000000 0.000000
9 C 2 Y 0.000000 0.000000 0.000000 -0.829983 0.000000
10 C 2 Z 0.000000 0.000000 0.000000 0.000000 0.116073
11 C 3 S 0.000000 0.000000 -0.065603 0.000000 0.076526
12 C 3 S 0.000000 0.000000 0.400317 0.000000 -0.495092
13 C 3 X 0.000000 0.000000 -0.141269 0.000000 0.156417
14 C 3 Y 0.579618 0.579839 0.000000 -0.218079 0.000000
15 C 3 Z 0.000000 0.000000 -0.393426 0.000000 0.282686
16 C 4 S 0.000000 0.000000 0.065603 0.000000 -0.076526
17 C 4 S 0.000000 0.000000 -0.400317 0.000000 0.495092
18 C 4 X 0.000000 0.000000 -0.141269 0.000000 0.156417
19 C 4 Y -0.579618 0.579839 0.000000 0.218079 0.000000
20 C 4 Z 0.000000 0.000000 -0.393426 0.000000 0.282686
21 C 5 S 0.000000 0.000000 -0.065603 0.000000 -0.076526
22 C 5 S 0.000000 0.000000 0.400317 0.000000 0.495092
23 C 5 X 0.000000 0.000000 -0.141269 0.000000 -0.156417
24 C 5 Y 0.579618 -0.579839 0.000000 0.218079 0.000000
25 C 5 Z 0.000000 0.000000 0.393426 0.000000 0.282686
26 C 6 S 0.000000 0.000000 0.065603 0.000000 0.076526
27 C 6 S 0.000000 0.000000 -0.400317 0.000000 -0.495092
28 C 6 X 0.000000 0.000000 -0.141269 0.000000 -0.156417
29 C 6 Y -0.579618 -0.579839 0.000000 -0.218079 0.000000
30 C 6 Z 0.000000 0.000000 0.393426 0.000000 0.282686
31 H 7 S 0.000000 0.000000 -0.216449 0.000000 0.297957
32 H 8 S 0.000000 0.000000 0.216449 0.000000 -0.297957
33 H 9 S 0.000000 0.000000 -0.216449 0.000000 -0.297957
34 H 10 S 0.000000 0.000000 0.216449 0.000000 0.297957
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -658.4193199139
TWO ELECTRON ENERGY = 253.4241652399
NUCLEAR REPULSION ENERGY = 176.3284658827
------------------
TOTAL ENERGY = -228.6666887912
ELECTRON-ELECTRON POTENTIAL ENERGY = 253.4241652399
NUCLEUS-ELECTRON POTENTIAL ENERGY = -887.3462082935
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 176.3284658827
------------------
TOTAL POTENTIAL ENERGY = -457.5935771709
TOTAL KINETIC ENERGY = 228.9268883796
VIRIAL RATIO (V/T) = 1.9988633944
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -151.5709911559
BARE H ENERGY= -658.4193199139
ELECTRONIC ENERGY = -404.9951555349
KINETIC ENERGY= 228.9268883796
N-N REPULSION= 176.3284658827
TOTAL ENERGY= -228.6666896522
SIGMA PART(1+2)= -367.4442409513
(K,V1,2)= 221.2641032112 -806.6971100285 217.9887658660
PI PART(1+2)= -37.5509145837
(K,V1,2)= 7.6627851684 -80.6490982650 35.4353985130
SIGMA SKELETON, ERROR= -191.1157750686 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 -0.000428 -0.000420 -0.000110 -0.000085 1.000460
2 -0.000428 -0.000420 -0.000110 -0.000085 1.000460
3 0.500349 0.500345 0.500177 0.500165 -0.000230
4 0.500349 0.500345 0.500177 0.500165 -0.000230
5 0.500349 0.500345 0.500177 0.500165 -0.000230
6 0.500349 0.500345 0.500177 0.500165 -0.000230
7 -0.000135 -0.000135 -0.000123 -0.000123 0.000000
8 -0.000135 -0.000135 -0.000123 -0.000123 0.000000
9 -0.000135 -0.000135 -0.000123 -0.000123 0.000000
10 -0.000135 -0.000135 -0.000123 -0.000123 0.000000
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 1.000425 0.325435 0.229596 0.719933 0.084692
2 1.000425 0.325435 0.229596 0.719933 0.084692
3 -0.000213 0.324429 0.368846 0.121423 0.291842
4 -0.000213 0.324429 0.368846 0.121423 0.291842
5 -0.000213 0.324429 0.368846 0.121423 0.291842
6 -0.000213 0.324429 0.368846 0.121423 0.291842
7 0.000000 0.012854 0.016356 0.018611 0.165812
8 0.000000 0.012854 0.016356 0.018611 0.165812
9 0.000000 0.012854 0.016356 0.018611 0.165812
10 0.000000 0.012854 0.016356 0.018611 0.165812
11 12 13 14 15
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.034545 0.107539 0.145505 0.134486 0.503932
2 0.034545 0.107539 0.145505 0.134486 0.503932
3 0.299663 0.289460 0.262409 0.385663 0.248034
4 0.299663 0.289460 0.262409 0.385663 0.248034
5 0.299663 0.289460 0.262409 0.385663 0.248034
6 0.299663 0.289460 0.262409 0.385663 0.248034
7 0.183064 0.156770 0.164839 0.047094 0.000000
8 0.183064 0.156770 0.164839 0.047094 0.000000
9 0.183064 0.156770 0.164839 0.047094 0.000000
10 0.183064 0.156770 0.164839 0.047094 0.000000
16 17 18 19 20
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.548166 0.230866 0.111434 0.358683 0.496068
2 0.548166 0.230866 0.111434 0.358683 0.496068
3 0.163004 0.360342 0.444283 0.264918 0.251966
4 0.163004 0.360342 0.444283 0.264918 0.251966
5 0.163004 0.360342 0.444283 0.264918 0.251966
6 0.163004 0.360342 0.444283 0.264918 0.251966
7 0.062913 0.024224 0.000000 0.055741 0.000000
8 0.062913 0.024224 0.000000 0.055741 0.000000
9 0.062913 0.024224 0.000000 0.055741 0.000000
10 0.062913 0.024224 0.000000 0.055741 0.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 C 1 S 1.99575 1.98903
2 C 1 S 1.01423 0.95239
3 C 1 X 1.02589 1.06648
4 C 1 Y 1.11143 1.11339
5 C 1 Z 0.88342 0.92870
6 C 2 S 1.99575 1.98903
7 C 2 S 1.01423 0.95239
8 C 2 X 1.02589 1.06648
9 C 2 Y 1.11143 1.11339
10 C 2 Z 0.88342 0.92870
11 C 3 S 1.99597 1.98997
12 C 3 S 1.12313 1.01762
13 C 3 X 1.00080 1.03679
14 C 3 Y 0.94428 0.94330
15 C 3 Z 1.01270 1.04372
16 C 4 S 1.99597 1.98997
17 C 4 S 1.12313 1.01762
18 C 4 X 1.00080 1.03679
19 C 4 Y 0.94428 0.94330
20 C 4 Z 1.01270 1.04372
21 C 5 S 1.99597 1.98997
22 C 5 S 1.12313 1.01762
23 C 5 X 1.00080 1.03679
24 C 5 Y 0.94428 0.94330
25 C 5 Z 1.01270 1.04372
26 C 6 S 1.99597 1.98997
27 C 6 S 1.12313 1.01762
28 C 6 X 1.00080 1.03679
29 C 6 Y 0.94428 0.94330
30 C 6 Z 1.01270 1.04372
31 H 7 S 0.90776 0.94360
32 H 8 S 0.90776 0.94360
33 H 9 S 0.90776 0.94360
34 H 10 S 0.90776 0.94360
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 4.8443462
2 0.6348494 4.8443462
3 0.2991656 -0.0150282 4.8925665
4 -0.0150282 0.2991656 0.0009359 4.8925665
5 -0.0150282 0.2991656 -0.0011833 0.5249316 4.8925665
6 0.2991656 -0.0150282 0.5249316 -0.0011833 0.0009359
7 -0.0081504 -0.0002234 0.3867544 -0.0000231 0.0000763
8 -0.0002234 -0.0081504 -0.0000231 0.3867544 -0.0113204
9 -0.0002234 -0.0081504 0.0000763 -0.0113204 0.3867544
10 -0.0081504 -0.0002234 -0.0113204 0.0000763 -0.0000231
6 7 8 9 10
6 4.8925665
7 -0.0113204 0.5414061
8 0.0000763 0.0000008 0.5414061
9 -0.0000231 -0.0000057 -0.0007511 0.5414061
10 0.3867544 -0.0007511 -0.0000057 0.0000008 0.5414061
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 C1 6.030723 -0.030723 6.049988 -0.049988
2 C1 6.030723 -0.030723 6.049988 -0.049988
3 C3 6.076875 -0.076875 6.031404 -0.031404
4 C3 6.076875 -0.076875 6.031404 -0.031404
5 C3 6.076875 -0.076875 6.031404 -0.031404
6 C3 6.076875 -0.076875 6.031404 -0.031404
7 H7 0.907763 0.092237 0.943602 0.056398
8 H7 0.907763 0.092237 0.943602 0.056398
9 H7 0.907763 0.092237 0.943602 0.056398
10 H7 0.907763 0.092237 0.943602 0.056398
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.282 1.824 1 3 1.460 0.999 1 4 2.670 0.055
1 5 2.670 0.055 1 6 1.460 0.999 2 3 2.670 0.055
2 4 1.460 0.999 2 5 1.460 0.999 2 6 2.670 0.055
3 6 1.296 1.904 3 7 1.073 0.959 4 5 1.296 1.904
4 8 1.073 0.959 5 9 1.073 0.959 6 10 1.073 0.959
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 C1 3.966 3.966 0.000
2 C1 3.966 3.966 0.000
3 C3 3.964 3.964 0.000
4 C3 3.964 3.964 0.000
5 C3 3.964 3.964 0.000
6 C3 3.964 3.964 0.000
7 H7 0.991 0.991 0.000
8 H7 0.991 0.991 0.000
9 H7 0.991 0.991 0.000
10 H7 0.991 0.991 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 0.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 0.000000 0.000000
...... END OF PROPERTY EVALUATION ......
Input
!
! Bicyclopropylium
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=+2 RUNTYP=OPTIMIZE
COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
Bicyclopropylium
CNH 2
C1
C2 1 1.40
C3 2 1.40 1 150.00
C4 2 1.40 1 150.00 3 180.00 0
C5 1 1.40 2 150.00 3 180.00 0
C6 1 1.40 2 150.00 3 0.00 0
H7 3 1.10 2 150.00 4 180.00 0
H8 4 1.10 2 150.00 3 180.00 0
H9 5 1.10 1 150.00 6 180.00 0
H10 6 1.10 1 150.00 5 180.00 0
$END
Output
THE MOMENTS OF INERTIA ARE (AMU-ANGSTROM**2)
IXX= 211.760 IYY= 246.290 IZZ= 34.530
ATOM ATOMIC
COORDINATES (BOHR)
CHARGE
X
Y Z
C1 6.0 0.0000000000
0.0000000000 1.3228081914
C1 6.0 0.0000000000
0.0000000000 -1.3228081914
C3 6.0 -1.3228081914
0.0000000000 3.6139791876
C3 6.0 1.3228081914
0.0000000000 -3.6139791876
C3 6.0 -1.3228081914
0.0000000000 -3.6139791876
C3 6.0 1.3228081914
0.0000000000 3.6139791876
H7 1.0 -3.1230139741
0.0000000000 4.6533284808
H7 1.0 3.1230139741
0.0000000000 -4.6533284808
H7 1.0 -3.1230139741
0.0000000000 -4.6533284808
H7 1.0 3.1230139741
0.0000000000 4.6533284808
ATOMIC BASIS SET
----------------
THE CONTRACTED PRIMITIVE FUNCTIONS HAVE BEEN UNNORMALIZED
THE CONTRACTED BASIS FUNCTIONS ARE NOW NORMALIZED TO UNITY
SHELL TYPE PRIM EXPONENT CONTRACTION COEFFICIENTS
C1
3 S 1 742.737049 0.929185 ( 0.009164)
3 S 2 136.180025 1.402437 ( 0.049361)
3 S 3 38.098264 1.841991 ( 0.168538)
3 S 4 13.087782 1.817278 ( 0.370563)
3 S 5 5.082369 1.004768 ( 0.416492)
3 S 6 2.093200 0.161650 ( 0.130334)
4 L 7 30.497240 -0.122578 ( -0.013253) 0.384077 ( 0.003760)
4 L 8 6.036200 -0.128975 ( -0.046992) 0.508157 ( 0.037679)
4 L 9 1.876046 -0.038599 ( -0.033785) 0.544234 ( 0.173897)
4 L 10 0.721783 0.139661 ( 0.250242) 0.396426 ( 0.418036)
4 L 11 0.313471 0.177689 ( 0.595117) 0.142381 ( 0.425860)
4 L 12 0.143687 0.040037 ( 0.240706) 0.012825 ( 0.101708)
TOTAL NUMBER OF SHELLS = 16
TOTAL NUMBER OF BASIS FUNCTIONS = 34
NUMBER OF ELECTRONS = 38
CHARGE OF MOLECULE = 2
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 19
NUMBER OF OCCUPIED ORBITALS (BETA ) = 19
TOTAL NUMBER OF ATOMS = 10
THE NUCLEAR REPULSION ENERGY IS 174.4674443816
1NSERCH= 0
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X
Y
Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000
-0.7000000000
C3 6.0 0.7000000000 0.0000000000
1.9124355653
H7 1.0 1.6526279442 0.0000000000
2.4624355653
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X
Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000
0.7000000000
C1 6.0 0.0000000000 0.0000000000
-0.7000000000
C3 6.0 -0.7000000000 0.0000000000
1.9124355653
C3 6.0 0.7000000000 0.0000000000
-1.9124355653
C3 6.0 -0.7000000000 0.0000000000
-1.9124355653
C3 6.0 0.7000000000 0.0000000000
1.9124355653
H7 1.0 -1.6526279442 0.0000000000
2.4624355653
H7 1.0 1.6526279442 0.0000000000
-2.4624355653
H7 1.0 -1.6526279442 0.0000000000
-2.4624355653
H7 1.0 1.6526279442 0.0000000000
2.4624355653
THE CURRENT FULLY SUBSTITUTED Z-MATRIX IS
C1
C1 1 1.4000000
C3 2 1.4000000 1 150.0000000
C3 2 1.4000000 1 150.0000000 3 180.0000000 0
C3 1 1.4000000 2 150.0000000 3 180.0000000 0
C3 1 1.4000000 2 150.0000000 3 0.0000000 0
H7 3 1.1000000 2 150.0000000 4 180.0000000 0
H7 4 1.1000000 2 150.0000000 3 180.0000000 0
H7 5 1.1000000 1 150.0000000 6 180.0000000 0
H7 6 1.1000000 1 150.0000000 5 180.0000000 0
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 174.4674443816
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=T
DENSITY CONV= 1.00E-05
SOSCF WILL OPTIMIZE 285 ORBITAL ROTATIONS, SOGTOL= 0.250
MEMORY REQUIRED FOR RHF STEP= 17205 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE ORB. GRAD
1 0 0 -227.717029048 -227.717029048 0.320557993 0.000000000
---------------START SECOND ORDER SCF---------------
2 1 0 -228.146752865 -0.429723816 0.098062969 0.032251824
3 2 0 -228.156583644 -0.009830779 0.034757742 0.021210645
4 3 0 -228.160389937 -0.003806293 0.004565201 0.002074736
5 4 0 -228.160457591 -0.000067654 0.000901049 0.000477604
6 5 0 -228.160460289 -0.000002698 0.000142158 0.000057738
7 6 0 -228.160460330 -0.000000042 0.000072666 0.000019181
8 7 0 -228.160460336 -0.000000006 0.000017319 0.000005943
9 8 0 -228.160460336 0.000000000 0.000008250 0.000001867
10 9 0 -228.160460336 0.000000000 0.000000879 0.000000330
-----------------
DENSITY CONVERGED
-----------------
TIME TO FORM FOCK OPERATORS= 0.0 SECONDS ( 0.0 SEC/ITER)
TIME TO SOLVE SCF EQUATIONS= 0.0 SECONDS ( 0.0 SEC/ITER)
FINAL ENERGY IS -228.1604603363 AFTER 10
ITERATIONS
------------
EIGENVECTORS
------------
HOMO-1
HOMO
LUMO
16 17
18 19
20
-0.9686
-0.9368 -0.9019 -0.8776
-0.3404
BU AG
BG BG
BU
1 C 1 S 0.000000 -0.006193 0.000000
0.000000 0.000000
2 C 1 S 0.000000 0.074490
0.000000 0.000000 0.000000
3 C 1 X 0.000000 0.000000
0.397806 0.000000 0.000000
4 C 1 Y 0.417196 0.000000
0.000000 0.340079 0.518623
5 C 1 Z 0.000000 0.308660
0.000000 0.000000 0.000000
6 C 2 S 0.000000 -0.006193 0.000000
0.000000 0.000000
7 C 2 S 0.000000 0.074490
0.000000 0.000000 0.000000
8 C 2 X 0.000000 0.000000 -0.397806
0.000000 0.000000
9 C 2 Y 0.417196 0.000000
0.000000 -0.340079 0.518623
10 C 2 Z 0.000000 -0.308660 0.000000
0.000000 0.000000
11 C 3 S 0.000000 0.006173 0.017543
0.000000 0.000000
12 C 3 S 0.000000 -0.042119 -0.099345
0.000000 0.000000
13 C 3 X 0.000000 -0.302995 -0.108593
0.000000 0.000000
14 C 3 Y 0.276098 0.000000 0.000000
0.358677 -0.380868
15 C 3 Z 0.000000 -0.236768 0.312965
0.000000 0.000000
16 C 4 S 0.000000 0.006173 0.017543
0.000000 0.000000
17 C 4 S 0.000000 -0.042119 -0.099345
0.000000 0.000000
18 C 4 X 0.000000 0.302995 0.108593
0.000000 0.000000
19 C 4 Y 0.276098 0.000000 0.000000
-0.358677 -0.380868
20 C 4 Z 0.000000 0.236768 -0.312965
0.000000 0.000000
21 C 5 S 0.000000 0.006173 -0.017543
0.000000 0.000000
22 C 5 S 0.000000 -0.042119 0.099345
0.000000 0.000000
23 C 5 X 0.000000 -0.302995 0.108593
0.000000 0.000000
24 C 5 Y 0.276098 0.000000 0.000000
-0.358677 -0.380868
25 C 5 Z 0.000000 0.236768 0.312965
0.000000 0.000000
26 C 6 S 0.000000 0.006173 -0.017543
0.000000 0.000000
27 C 6 S 0.000000 -0.042119 0.099345
0.000000 0.000000
28 C 6 X 0.000000 0.302995 -0.108593
0.000000 0.000000
29 C 6 Y 0.276098 0.000000 0.000000
0.358677 -0.380868
30 C 6 Z 0.000000 -0.236768 -0.312965
0.000000 0.000000
31 H 7 S 0.000000 0.092209 0.160113
0.000000 0.000000
32 H 8 S 0.000000 0.092209 0.160113
0.000000 0.000000
33 H 9 S 0.000000 0.092209 -0.160113
0.000000 0.000000
34 H 10 S 0.000000 0.092209 -0.160113
0.000000 0.000000
HOMO-1
von vorn
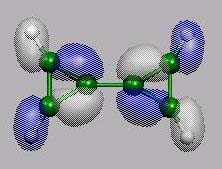
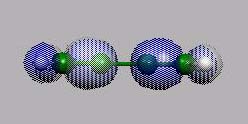
HOMO
von vorn
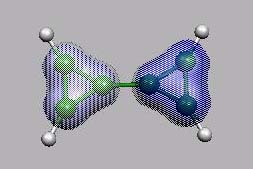
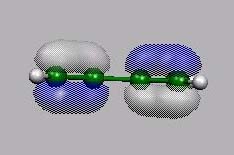
LUMO
von vorn
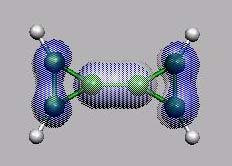
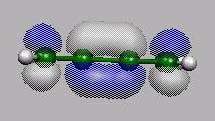
21 22 23 24 25
-0.2112 -0.2112 -0.0973 -0.0530 0.0447
AG AU BG BU AU
1 C 1 S 0.000000 0.000000 0.000000 0.000000 -0.180594
2 C 1 S 0.000000 0.000000 0.000000 0.000000 1.191098
3 C 1 X 0.000000 0.000000 0.000000 0.564874 0.000000
4 C 1 Y 0.000000 0.000000 0.759567 0.000000 0.000000
5 C 1 Z 0.000000 0.000000 0.000000 0.000000 -0.169511
6 C 2 S 0.000000 0.000000 0.000000 0.000000 0.180594
7 C 2 S 0.000000 0.000000 0.000000 0.000000 -1.191098
8 C 2 X 0.000000 0.000000 0.000000 0.564874 0.000000
9 C 2 Y 0.000000 0.000000 -0.759567 0.000000 0.000000
10 C 2 Z 0.000000 0.000000 0.000000 0.000000 -0.169511
11 C 3 S 0.000000 0.000000 0.000000 -0.053156 0.060311
12 C 3 S 0.000000 0.000000 0.000000 0.343643 -0.371984
13 C 3 X 0.000000 0.000000 0.000000 -0.135691 0.052704
14 C 3 Y 0.563283 0.563561 -0.310659 0.000000 0.000000
15 C 3 Z 0.000000 0.000000 0.000000 -0.446686 0.337984
16 C 4 S 0.000000 0.000000 0.000000 0.053156 -0.060311
17 C 4 S 0.000000 0.000000 0.000000 -0.343643 0.371984
18 C 4 X 0.000000 0.000000 0.000000 -0.135691 0.052704
19 C 4 Y -0.563283 0.563561 0.310659 0.000000 0.000000
20 C 4 Z 0.000000 0.000000 0.000000 -0.446686 0.337984
21 C 5 S 0.000000 0.000000 0.000000 -0.053156 -0.060311
22 C 5 S 0.000000 0.000000 0.000000 0.343643 0.371984
23 C 5 X 0.000000 0.000000 0.000000 -0.135691 -0.052704
24 C 5 Y 0.563283 -0.563561 0.310659 0.000000 0.000000
25 C 5 Z 0.000000 0.000000 0.000000 0.446686 0.337984
26 C 6 S 0.000000 0.000000 0.000000 0.053156 0.060311
27 C 6 S 0.000000 0.000000 0.000000 -0.343643 -0.371984
28 C 6 X 0.000000 0.000000 0.000000 -0.135691 -0.052704
29 C 6 Y -0.563283 -0.563561 -0.310659 0.000000 0.000000
30 C 6 Z 0.000000 0.000000 0.000000 0.446686 0.337984
31 H 7 S 0.000000 0.000000 0.000000 -0.163794 0.139235
32 H 8 S 0.000000 0.000000 0.000000 0.163794 -0.139235
33 H 9 S 0.000000 0.000000 0.000000 -0.163794 -0.139235
34 H 10 S 0.000000 0.000000 0.000000 0.163794 0.139235
LUMO+1
von vorn
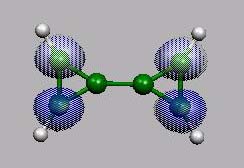

------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -633.6696609515
TWO ELECTRON ENERGY = 231.0417562335
NUCLEAR REPULSION ENERGY = 174.4674443816
------------------
TOTAL ENERGY = -228.1604603363
ELECTRON-ELECTRON POTENTIAL ENERGY = 231.0417562335
NUCLEUS-ELECTRON POTENTIAL ENERGY = -859.9679755189
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 174.4674443816
------------------
TOTAL POTENTIAL ENERGY = -454.4587749038
TOTAL KINETIC ENERGY = 226.2983145675
VIRIAL RATIO (V/T) = 2.0082287213
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -171.5861455947
BARE H ENERGY= -633.6696609515
ELECTRONIC ENERGY = -402.6279032731
KINETIC ENERGY= 226.2983145675
N-N REPULSION= 174.4674443816
TOTAL ENERGY= -228.1604588915
SIGMA PART(1+2)= -376.4304963859
(K,V1,2)= 221.7348320078 -806.7021374910 208.5368090973
PI PART(1+2)= -26.1974068871
(K,V1,2)= 4.5634825597 -53.2658380280 22.5049485811
SIGMA SKELETON, ERROR= -201.9630520043 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.997990 0.997691 0.002375 0.002120 -0.000153
2 0.997990 0.997691 0.002375 0.002120 -0.000153
3 0.001006 0.001155 0.498917 0.499045 0.500169
4 0.001006 0.001155 0.498917 0.499045 0.500169
5 0.001006 0.001155 0.498917 0.499045 0.500169
6 0.001006 0.001155 0.498917 0.499045 0.500169
7 -0.000001 -0.000001 -0.000105 -0.000105 -0.000093
8 -0.000001 -0.000001 -0.000105 -0.000105 -0.000093
9 -0.000001 -0.000001 -0.000105 -0.000105 -0.000093
10 -0.000001 -0.000001 -0.000105 -0.000105 -0.000093
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 -0.000127 0.388982 0.326453 0.785721 0.197707
2 -0.000127 0.388982 0.326453 0.785721 0.197707
3 0.500157 0.299027 0.329110 0.100290 0.306586
4 0.500157 0.299027 0.329110 0.100290 0.306586
5 0.500157 0.299027 0.329110 0.100290 0.306586
6 0.500157 0.299027 0.329110 0.100290 0.306586
7 -0.000093 0.006483 0.007664 0.006849 0.094560
8 -0.000093 0.006483 0.007664 0.006849 0.094560
9 -0.000093 0.006483 0.007664 0.006849 0.094560
10 -0.000093 0.006483 0.007664 0.006849 0.094560
11 12 13 14 15
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.089662 0.086440 0.106529 0.420650 0.114318
2 0.089662 0.086440 0.106529 0.420650 0.114318
3 0.333984 0.331146 0.306990 0.200005 0.399639
4 0.333984 0.331146 0.306990 0.200005 0.399639
5 0.333984 0.331146 0.306990 0.200005 0.399639
6 0.333984 0.331146 0.306990 0.200005 0.399639
7 0.121184 0.125634 0.139746 0.089671 0.043202
8 0.121184 0.125634 0.139746 0.089671 0.043202
9 0.121184 0.125634 0.139746 0.089671 0.043202
10 0.121184 0.125634 0.139746 0.089671 0.043202
16 17 18 19
2.000000 2.000000 2.000000 2.000000
1 0.525996 0.188335 0.359044 0.279429
2 0.525996 0.188335 0.359044 0.279429
3 0.237002 0.381732 0.251152 0.360285
4 0.237002 0.381732 0.251152 0.360285
5 0.237002 0.381732 0.251152 0.360285
6 0.237002 0.381732 0.251152 0.360285
7 0.000000 0.024101 0.069327 0.000000
8 0.000000 0.024101 0.069327 0.000000
9 0.000000 0.024101 0.069327 0.000000
10 0.000000 0.024101 0.069327 0.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 C 1 S 1.99594 1.98933
2 C 1 S 1.07193 0.99230
3 C 1 X 1.06678 1.10953
4 C 1 Y 0.80542 0.79964
5 C 1 Z 0.92909 0.95907
6 C 2 S 1.99594 1.98933
7 C 2 S 1.07193 0.99230
8 C 2 X 1.06678 1.10953
9 C 2 Y 0.80542 0.79964
10 C 2 Z 0.92909 0.95907
11 C 3 S 1.99630 1.99025
12 C 3 S 1.19845 1.08143
13 C 3 X 1.04259 1.05253
14 C 3 Y 0.59729 0.60018
15 C 3 Z 1.00276 1.04537
16 C 4 S 1.99630 1.99025
17 C 4 S 1.19845 1.08143
18 C 4 X 1.04259 1.05253
19 C 4 Y 0.59729 0.60018
20 C 4 Z 1.00276 1.04537
21 C 5 S 1.99630 1.99025
22 C 5 S 1.19845 1.08143
23 C 5 X 1.04259 1.05253
24 C 5 Y 0.59729 0.60018
25 C 5 Z 1.00276 1.04537
26 C 6 S 1.99630 1.99025
27 C 6 S 1.19845 1.08143
28 C 6 X 1.04259 1.05253
29 C 6 Y 0.59729 0.60018
30 C 6 Z 1.00276 1.04537
31 H 7 S 0.72802 0.80530
32 H 8 S 0.72802 0.80530
33 H 9 S 0.72802 0.80530
34 H 10 S 0.72802 0.80530
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 4.6970428
2 0.4159683 4.6970428
3 0.3956647 -0.0097895 4.6999660
4 -0.0097895 0.3956647 0.0006928 4.6999660
5 -0.0097895 0.3956647 -0.0010273 0.3881495 4.6999660
6 0.3956647 -0.0097895 0.3881495 -0.0010273 0.0006928
7 -0.0075282 -0.0002715 0.3707851 -0.0000138 0.0000556
8 -0.0002715 -0.0075282 -0.0000138 0.3707851 -0.0070863
9 -0.0002715 -0.0075282 0.0000556 -0.0070863 0.3707851
10 -0.0075282 -0.0002715 -0.0070863 0.0000556 -0.0000138
6 7 8 9 10
6 4.6999660
7 -0.0070863 0.3724235
8 0.0000556 0.0000004 0.3724235
9 -0.0000138 -0.0000031 -0.0003397 0.3724235
10 0.3707851 -0.0003397 -0.0000031 0.0000004 0.3724235
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 C1 5.869162 0.130838 5.849874 0.150126
2 C1 5.869162 0.130838 5.849874 0.150126
3 C3 5.837397 0.162603 5.769768 0.230232
4 C3 5.837397 0.162603 5.769768 0.230232
5 C3 5.837397 0.162603 5.769768 0.230232
6 C3 5.837397 0.162603 5.769768 0.230232
7 H7 0.728022 0.271978 0.805296 0.194704
8 H7 0.728022 0.271978 0.805296 0.194704
9 H7 0.728022 0.271978 0.805296 0.194704
10 H7 0.728022 0.271978 0.805296 0.194704
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.400 1.046 1 3 1.400 1.427 1 6 1.400 1.427
2 4 1.400 1.427 2 5 1.400 1.427 3 6 1.400 1.344
3 7 1.100 0.901 4 5 1.400 1.344 4 8 1.100 0.901
5 9 1.100 0.901 6 10 1.100 0.901
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 C1 3.947 3.947 0.000
2 C1 3.947 3.947 0.000
3 C3 3.740 3.740 0.000
4 C3 3.740 3.740 0.000
5 C3 3.740 3.740 0.000
6 C3 3.740 3.740 0.000
7 H7 0.926 0.926 0.000
8 H7 0.926 0.926 0.000
9 H7 0.926 0.926 0.000
10 H7 0.926 0.926 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 2.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 0.000000 0.000000
...... END OF PROPERTY EVALUATION ......
NSERCH= 12 ENERGY= -228.1680490
-----------------------
GRADIENT (HARTREE/BOHR)
-----------------------
ATOM ZNUC DE/DX DE/DY DE/DZ
--------------------------------------------------------------
1 C1 6.0 0.0000000 0.0000000 -0.0000118
2 C1 6.0 0.0000000 0.0000000 0.0000118
3 C3 6.0 -0.0000005 0.0000000 0.0000070
4 C3 6.0 0.0000005 0.0000000 -0.0000070
5 C3 6.0 -0.0000005 0.0000000 -0.0000070
6 C3 6.0 0.0000005 0.0000000 0.0000070
7 H7 1.0 -0.0000003 0.0000000 0.0000044
8 H7 1.0 0.0000003 0.0000000 -0.0000044
9 H7 1.0 -0.0000003 0.0000000 -0.0000044
10 H7 1.0 0.0000003 0.0000000 0.0000044
MAXIMUM GRADIENT = 0.0000118 RMS GRADIENT = 0.0000043
1 ***** EQUILIBRIUM GEOMETRY LOCATED
*****
Bicyclopropylium
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X
Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000
-0.7415772423
C3 6.0 0.7007235051 0.0000000000
1.9284932529
H7 1.0 1.6506485417 0.0000000000
2.4889544782
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000 0.7415772423
C1 6.0 0.0000000000 0.0000000000 -0.7415772423
C3 6.0 -0.7007235051 0.0000000000 1.9284932529
C3 6.0 0.7007235051 0.0000000000 -1.9284932529
C3 6.0 -0.7007235051 0.0000000000 -1.9284932529
C3 6.0 0.7007235051 0.0000000000 1.9284932529
H7 1.0 -1.6506485417 0.0000000000 2.4889544782
H7 1.0 1.6506485417 0.0000000000 -2.4889544782
H7 1.0 -1.6506485417 0.0000000000 -2.4889544782
H7 1.0 1.6506485417 0.0000000000 2.4889544782
THE CURRENT FULLY SUBSTITUTED Z-MATRIX IS
C1
C1 1 1.4831545
C3 2 1.3783262 1 149.4435327
C3 2 1.3783262 1 149.4435327 3 180.0000000 0
C3 1 1.3783262 2 149.4435327 3 180.0000000 0
C3 1 1.3783262 2 149.4435327 3 0.0000000 0
H7 3 1.1029390 2 151.0973162 4 180.0000000 0
H7 4 1.1029390 2 151.0973162 3 180.0000000 0
H7 5 1.1029390 1 151.0973162 6 180.0000000 0
H7 6 1.1029390 1 151.0973162 5 180.0000000 0
NUCLEAR ENERGY = 173.6963334365
ELECTRONIC ENERGY = -401.8643824025
TOTAL ENERGY = -228.1680489660
------------------
MOLECULAR ORBITALS
------------------
16 17 18 19 20
-0.9656 -0.9265 -0.9178 -0.8902 -0.3158
BU AG BG BG BU
1 C 1 S 0.000000 -0.003477 0.000000 0.000000 0.000000
2 C 1 S 0.000000 0.053851 0.000000 0.000000 0.000000
3 C 1 X 0.000000 0.000000 0.390210 0.000000 0.000000
4 C 1 Y 0.408589 0.000000 0.000000 0.348035 0.539023
5 C 1 Z 0.000000 0.317665 0.000000 0.000000 0.000000
6 C 2 S 0.000000 -0.003477 0.000000 0.000000 0.000000
7 C 2 S 0.000000 0.053851 0.000000 0.000000 0.000000
8 C 2 X 0.000000 0.000000 -0.390210 0.000000 0.000000
9 C 2 Y 0.408589 0.000000 0.000000 -0.348035 0.539023
10 C 2 Z 0.000000 -0.317665 0.000000 0.000000 0.000000
11 C 3 S 0.000000 0.004760 0.014775 0.000000 0.000000
12 C 3 S 0.000000 -0.040371 -0.085353 0.000000 0.000000
13 C 3 X 0.000000 -0.300037 -0.115154 0.000000 0.000000
14 C 3 Y 0.283467 0.000000 0.000000 0.351097 -0.377565
15 C 3 Z 0.000000 -0.240014 0.308288 0.000000 0.000000
16 C 4 S 0.000000 0.004760 0.014775 0.000000 0.000000
17 C 4 S 0.000000 -0.040371 -0.085353 0.000000 0.000000
18 C 4 X 0.000000 0.300037 0.115154 0.000000 0.000000
19 C 4 Y 0.283467 0.000000 0.000000 -0.351097 -0.377565
20 C 4 Z 0.000000 0.240014 -0.308288 0.000000 0.000000
21 C 5 S 0.000000 0.004760 -0.014775 0.000000 0.000000
22 C 5 S 0.000000 -0.040371 0.085353 0.000000 0.000000
23 C 5 X 0.000000 -0.300037 0.115154 0.000000 0.000000
24 C 5 Y 0.283467 0.000000 0.000000 -0.351097 -0.377565
25 C 5 Z 0.000000 0.240014 0.308288 0.000000 0.000000
26 C 6 S 0.000000 0.004760 -0.014775 0.000000 0.000000
27 C 6 S 0.000000 -0.040371 0.085353 0.000000 0.000000
28 C 6 X 0.000000 0.300037 -0.115154 0.000000 0.000000
29 C 6 Y 0.283467 0.000000 0.000000 0.351097 -0.377565
30 C 6 Z 0.000000 -0.240014 -0.308288 0.000000 0.000000
31 H 7 S 0.000000 0.090916 0.162621 0.000000 0.000000
32 H 8 S 0.000000 0.090916 0.162621 0.000000 0.000000
33 H 9 S 0.000000 0.090916 -0.162621 0.000000 0.000000
34 H 10 S 0.000000 0.090916 -0.162621 0.000000 0.000000
21 22 23 24 25
-0.2122 -0.2120 -0.1071 -0.0258 0.0087
AG AU BG BU AU
1 C 1 S 0.000000 0.000000 0.000000 0.000000 -0.181450
2 C 1 S 0.000000 0.000000 0.000000 0.000000 1.166358
3 C 1 X 0.000000 0.000000 0.000000 0.583654 0.000000
4 C 1 Y 0.000000 0.000000 0.741376 0.000000 0.000000
5 C 1 Z 0.000000 0.000000 0.000000 0.000000 -0.324923
6 C 2 S 0.000000 0.000000 0.000000 0.000000 0.181450
7 C 2 S 0.000000 0.000000 0.000000 0.000000 -1.166358
8 C 2 X 0.000000 0.000000 0.000000 0.583654 0.000000
9 C 2 Y 0.000000 0.000000 -0.741376 0.000000 0.000000
10 C 2 Z 0.000000 0.000000 0.000000 0.000000 -0.324923
11 C 3 S 0.000000 0.000000 0.000000 -0.049603 0.047553
12 C 3 S 0.000000 0.000000 0.000000 0.327908 -0.272763
13 C 3 X 0.000000 0.000000 0.000000 -0.160233 0.025373
14 C 3 Y 0.563094 0.563348 -0.321399 0.000000 0.000000
15 C 3 Z 0.000000 0.000000 0.000000 -0.446912 0.321610
16 C 4 S 0.000000 0.000000 0.000000 0.049603 -0.047553
17 C 4 S 0.000000 0.000000 0.000000 -0.327908 0.272763
18 C 4 X 0.000000 0.000000 0.000000 -0.160233 0.025373
19 C 4 Y -0.563094 0.563348 0.321399 0.000000 0.000000
20 C 4 Z 0.000000 0.000000 0.000000 -0.446912 0.321610
21 C 5 S 0.000000 0.000000 0.000000 -0.049603 -0.047553
22 C 5 S 0.000000 0.000000 0.000000 0.327908 0.272763
23 C 5 X 0.000000 0.000000 0.000000 -0.160233 -0.025373
24 C 5 Y 0.563094 -0.563348 0.321399 0.000000 0.000000
25 C 5 Z 0.000000 0.000000 0.000000 0.446912 0.321610
26 C 6 S 0.000000 0.000000 0.000000 0.049603 0.047553
27 C 6 S 0.000000 0.000000 0.000000 -0.327908 -0.272763
28 C 6 X 0.000000 0.000000 0.000000 -0.160233 -0.025373
29 C 6 Y -0.563094 -0.563348 -0.321399 0.000000 0.000000
30 C 6 Z 0.000000 0.000000 0.000000 0.446912 0.321610
31 H 7 S 0.000000 0.000000 0.000000 -0.162242 0.051682
32 H 8 S 0.000000 0.000000 0.000000 0.162242 -0.051682
33 H 9 S 0.000000 0.000000 0.000000 -0.162242 -0.051682
34 H 10 S 0.000000 0.000000 0.000000 0.162242 0.051682
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -632.1540737757
TWO ELECTRON ENERGY = 230.2896913732
NUCLEAR REPULSION ENERGY = 173.6963334365
------------------
TOTAL ENERGY = -228.1680489660
ELECTRON-ELECTRON POTENTIAL ENERGY = 230.2896913732
NUCLEUS-ELECTRON POTENTIAL ENERGY = -858.4445385996
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 173.6963334365
------------------
TOTAL POTENTIAL ENERGY = -454.4585137900
TOTAL KINETIC ENERGY = 226.2904648239
VIRIAL RATIO (V/T) = 2.0082972305
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -171.5746935999
BARE H ENERGY= -632.1540737757
ELECTRONIC ENERGY = -401.8643836878
KINETIC ENERGY= 226.2904648239
N-N REPULSION= 173.6963334365
TOTAL ENERGY= -228.1680502513
SIGMA PART(1+2)= -375.7344636712
(K,V1,2)= 221.7357587088 -805.3416283729 207.8714059929
PI PART(1+2)= -26.1299200166
(K,V1,2)= 4.5547061152 -53.1029102268 22.4182840950
SIGMA SKELETON, ERROR= -202.0381302347 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.998544 0.998389 0.001710 0.001611 -0.000167
2 0.998544 0.998389 0.001710 0.001611 -0.000167
3 0.000729 0.000806 0.499247 0.499296 0.500175
4 0.000729 0.000806 0.499247 0.499296 0.500175
5 0.000729 0.000806 0.499247 0.499296 0.500175
6 0.000729 0.000806 0.499247 0.499296 0.500175
7 -0.000001 -0.000001 -0.000102 -0.000102 -0.000091
8 -0.000001 -0.000001 -0.000102 -0.000102 -0.000091
9 -0.000001 -0.000001 -0.000102 -0.000102 -0.000091
10 -0.000001 -0.000001 -0.000102 -0.000102 -0.000091
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 -0.000142 0.380637 0.337756 0.771874 0.198824
2 -0.000142 0.380637 0.337756 0.771874 0.198824
3 0.500162 0.303286 0.323913 0.105843 0.307757
4 0.500162 0.303286 0.323913 0.105843 0.307757
5 0.500162 0.303286 0.323913 0.105843 0.307757
6 0.500162 0.303286 0.323913 0.105843 0.307757
7 -0.000091 0.006396 0.007209 0.008220 0.092831
8 -0.000091 0.006396 0.007209 0.008220 0.092831
9 -0.000091 0.006396 0.007209 0.008220 0.092831
10 -0.000091 0.006396 0.007209 0.008220 0.092831
11 12 13 14 15
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.103778 0.081220 0.116630 0.398721 0.114396
2 0.103778 0.081220 0.116630 0.398721 0.114396
3 0.331889 0.334025 0.302741 0.208473 0.399657
4 0.331889 0.334025 0.302741 0.208473 0.399657
5 0.331889 0.334025 0.302741 0.208473 0.399657
6 0.331889 0.334025 0.302741 0.208473 0.399657
7 0.116222 0.125365 0.138944 0.092166 0.043145
8 0.116222 0.125365 0.138944 0.092166 0.043145
9 0.116222 0.125365 0.138944 0.092166 0.043145
10 0.116222 0.125365 0.138944 0.092166 0.043145
16 17 18 19
2.000000 2.000000 2.000000 2.000000
1 0.502400 0.200952 0.355027 0.300570
2 0.502400 0.200952 0.355027 0.300570
3 0.248800 0.376530 0.248628 0.349715
4 0.248800 0.376530 0.248628 0.349715
5 0.248800 0.376530 0.248628 0.349715
6 0.248800 0.376530 0.248628 0.349715
7 0.000000 0.022994 0.073859 0.000000
8 0.000000 0.022994 0.073859 0.000000
9 0.000000 0.022994 0.073859 0.000000
10 0.000000 0.022994 0.073859 0.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 C 1 S 1.99597 1.98917
2 C 1 S 1.09327 1.00811
3 C 1 X 1.05604 1.10296
4 C 1 Y 0.80297 0.79734
5 C 1 Z 0.91448 0.94567
6 C 2 S 1.99597 1.98917
7 C 2 S 1.09327 1.00811
8 C 2 X 1.05604 1.10296
9 C 2 Y 0.80297 0.79734
10 C 2 Z 0.91448 0.94567
11 C 3 S 1.99629 1.99031
12 C 3 S 1.19389 1.07647
13 C 3 X 1.04083 1.05015
14 C 3 Y 0.59851 0.60133
15 C 3 Z 1.01214 1.05620
16 C 4 S 1.99629 1.99031
17 C 4 S 1.19389 1.07647
18 C 4 X 1.04083 1.05015
19 C 4 Y 0.59851 0.60133
20 C 4 Z 1.01214 1.05620
21 C 5 S 1.99629 1.99031
22 C 5 S 1.19389 1.07647
23 C 5 X 1.04083 1.05015
24 C 5 Y 0.59851 0.60133
25 C 5 Z 1.01214 1.05620
26 C 6 S 1.99629 1.99031
27 C 6 S 1.19389 1.07647
28 C 6 X 1.04083 1.05015
29 C 6 Y 0.59851 0.60133
30 C 6 Z 1.01214 1.05620
31 H 7 S 0.72696 0.80391
32 H 8 S 0.72696 0.80391
33 H 9 S 0.72696 0.80391
34 H 10 S 0.72696 0.80391
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 4.6991208
2 0.3910430 4.6991208
3 0.4033377 -0.0093034 4.7025039
4 -0.0093034 0.4033377 0.0005554 4.7025039
5 -0.0093034 0.4033377 -0.0007232 0.3817251 4.7025039
6 0.4033377 -0.0093034 0.3817251 -0.0007232 0.0005554
7 -0.0074783 -0.0002728 0.3704953 -0.0000098 0.0000437
8 -0.0002728 -0.0074783 -0.0000098 0.3704953 -0.0069537
9 -0.0002728 -0.0074783 0.0000437 -0.0069537 0.3704953
10 -0.0074783 -0.0002728 -0.0069537 0.0000437 -0.0000098
6 7 8 9 10
6 4.7025039
7 -0.0069537 0.3714949
8 0.0000437 0.0000003 0.3714949
9 -0.0000098 -0.0000020 -0.0003535 0.3714949
10 0.3704953 -0.0003535 -0.0000020 0.0000003 0.3714949
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 C1 5.862730 0.137270 5.843244 0.156756
2 C1 5.862730 0.137270 5.843244 0.156756
3 C3 5.841671 0.158329 5.774469 0.225531
4 C3 5.841671 0.158329 5.774469 0.225531
5 C3 5.841671 0.158329 5.774469 0.225531
6 C3 5.841671 0.158329 5.774469 0.225531
7 H7 0.726964 0.273036 0.803909 0.196091
8 H7 0.726964 0.273036 0.803909 0.196091
9 H7 0.726964 0.273036 0.803909 0.196091
10 H7 0.726964 0.273036 0.803909 0.196091
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.483 1.002 1 3 1.378 1.444 1 6 1.378 1.444
2 4 1.378 1.444 2 5 1.378 1.444 3 6 1.401 1.340
3 7 1.103 0.902 4 5 1.401 1.340 4 8 1.103 0.902
5 9 1.103 0.902 6 10 1.103 0.902
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 C1 3.943 3.943 0.000
2 C1 3.943 3.943 0.000
3 C3 3.742 3.742 0.000
4 C3 3.742 3.742 0.000
5 C3 3.742 3.742 0.000
6 C3 3.742 3.742 0.000
7 H7 0.925 0.925 0.000
8 H7 0.925 0.925 0.000
9 H7 0.925 0.925 0.000
10 H7 0.925 0.925 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 2.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 0.000000 0.000000
...... END OF PROPERTY EVALUATION ......
| Startseite |