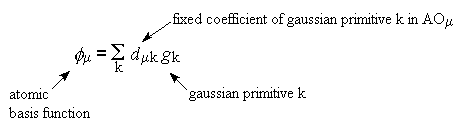Molekülorbitale werden im Allgemeinen durch eine Linearkombination von
Atomorbitalen dargestellt (LCAO-MO). Dabei werden die Atomorbitale durch eine
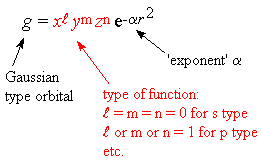
Summe von Gaussfunktionen gebildet (grafische Darstellung).
Der einfachste Basissatz wird als STO-3G (Slater Type Orbitals)
bezeichnet. Dabei werden die AO's durch 3 Gaussfunktionen gebildet. Minimaler
Basissatz bedeutet, dass nur so viele AO's verwendet werden, um die Elektronen des
neutralen Atoms unterzubringen.
Die entsprechende Befehlsgruppe im Input-File lautet:
GBASIS
= Name of the Gaussian basis set.
= MINI - Huzinaga's 3 gaussian
minimal basis set.
Available H-Rn.
= MIDI - Huzinaga's 21 split valence
basis set.
Available H-Xe.
= STO
- Pople's STO-NG minimal basis set.
Available H-Xe, for N=2,3,4,5,6
= N21 - Pople's N-21G split
valence basis set.
Available H-Xe, for NGAUSS=3.
Available H-Ar, for NGAUSS=6.
= N31
- Pople's N-31G split valence basis set.
Available H-Ne,P-Cl for NGAUSS=4.
Available H-He,C-F for NGAUSS=5.
Available H-Ar, for NGAUSS=6.
For Ga-Kr, N31 selects the BC basis.
= N311 - Pople's "triple split"
N-311G basis set.
Available H-Ne, for NGAUSS=6.
Selecting
N311 implies MC for Na-Ar.
= DZV
- "double zeta valence" basis set.
a synonym for DH for H,Li,Be-Ne,Al-Cl.
a synonym for BC for Ga-Kr.
= DH -
Dunning/Hay "double zeta" basis set.
(3s)/[2s] for H.
(9s,4p)/[3s,2p] for Li.
(9s,5p)/[3s,2p] for Be-Ne.
(11s,7p)/[6s,4p] for Al-Cl.
= BC -
Binning/Curtiss "double zeta" basis set.
(14s,11p,5d/[6s,4p,1d] for Ga-Kr.
= TZV
- "triple zeta valence" basis set.
(5s)/[3s] for H.
(10s,3p)/[4s,3p] for Li.
(10s,6p)/[5s,3p] for Be-Ne.
a synonym for MC for Na-Ar.
(14s,9p)/[8s,4p] for K-Ca.
(14s,11p,6d)/[10s,8p,3d] for Sc-Zn.
= MC
- McLean/Chandler "triple split"
basis.
(12s,9p)/[6s,5p] for Na-Ar.
Selecting MC implies 6-311G for H-Ne.
* * * the next two are ECP bases only * * *
GBASIS
= SBK - Stevens/Basch/Krauss/Jasien/Cundari
valence basis set, for Li-Rn. This
choice
implies an unscaled -31G basis for H-He.
= HW
- Hay/Wadt valence basis.
This is a -21 split, available Na-Xe,
except for the transition metals.
This implies a 3-21G basis for H-Ne.
* * * semiempirical basis sets * * *
The
elements for which these exist can be found
in the 'further information' section of this
manual. If you pick one of
these, all other data
in this group is ignored.
GBASIS
= MNDO - selects MNDO model hamiltonian
= AM1
- selects AM1 model hamiltonian
= PM3
- selects PM3 model hamiltonian
NGAUSS
= the number of Gaussians (N). This
parameter
pertains only to GBASIS=STO, N21, N31, or N311.
NDFUNC
= number of heavy atom polarization functions to
be used.
These are usually d functions, except
for MINI/MIDI.
The term "heavy" means Na on up
when GBASIS=STO, HW, or
N21, and from Li on up
otherwise.
The value may not exceed 3. The
variable POLAR selects
the actual exponents to
be used, see also SPLIT2
and SPLIT3. (default=0)
NFFUNC
= number of heavy atom f type polarization
functions to be used on
Li-Cl. This may only
be input as 0 or 1.
(default=0)
NPFUNC
= number of light atom, p type polarization
functions to be used on
H-He. This may not
exceed 3, see also
POLAR. (default=0)
DIFFSP
= flag to add diffuse sp (L) shell to heavy atoms.
Heavy means Li-F, Na-Cl,
Ga-Br, In-I, Tl-At.
The default is .FALSE.
DIFFS = flag to add diffuse s shell to hydrogens.
The default is .FALSE.
POLAR = exponent of polarization functions
= POPLE
(default for all other cases)
= POPN311
(default for GBASIS=N311, MC)
= DUNNING
(default for GBASIS=DH, DZV)
= HUZINAGA
(default for GBASIS=MINI, MIDI)
= HONDO7
(default for GBASIS=TZV)
SPLIT2
= an array of splitting factors used when NDFUNC
or NPFUNC is 2.
Default=2.0,0.5
SPLIT3 = an array of splitting factors used when NDFUNC
or NPFUNC is 3.
Default=4.00,1.00,0.25
==========================================================
The splitting factors are from the Pople school, and are
probably too far apart. See for
example the Binning and
Curtiss paper. For example, the
SPLIT2 value will usually cause an INCREASE over the 1d energy at the HF level for
hydrocarbons. The actual
exponents used for polarization functions, as well as for diffuse sp or s shells,
are described in the 'Further References' section of this manual.
This section also describes the sp part of the basis set chosen by
GBASIS fully, with all references cited.
Note that GAMESS always punches a full $DATA group.
Thus, if $BASIS does not quite
cover the basis you want, you can obtain this full $DATA group from EXETYP=CHECK,
and then change polarization exponents, add Rydbergs, etc.