Ungeradzahlige lineare Kette von H-Atomen
H32+
Input
!
! H3 2plus
!
$CONTRL SCFTYP=ROHF MULT=2 ICHARG=2 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=5000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H3 2plus
CNH 2
H 1.0 0.000 0.000 0.000
H 1.0 0.000 1.000 0.000
$END
Output
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
H 1.0 0.0000000000 0.0000000000 0.0000000000
H 1.0 0.0000000000 -1.8897259877 0.0000000000
H 1.0 0.0000000000 1.8897259877 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
H H H
1 H 0.0000000 1.0000000 * 1.0000000 *
2 H 1.0000000 * 0.0000000 2.0000000 *
3 H 1.0000000 * 2.0000000 * 0.0000000
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 1.3229431231
MAXIT = 30 NPUNCH= 2 MULT= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
MEMORY REQUIRED FOR UHF/ROHF STEP= 8126 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 -0.209307551 -0.209307551 0.158256476 0.000000000
2 1 -0.230397627 -0.021090076 0.061028503 0.000000000
3 2 -0.232985377 -0.002587750 0.021955840 0.000000000
4 3 -0.233302616 -0.000317239 0.007754881 0.000000000
5 0 -0.233341503 -0.000038887 0.004190677 0.000000000
6 1 -0.233346935 -0.000005433 0.000000846 0.000000000
7 2 -0.233346935 0.000000000 0.000000296 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -0.2333469352 AFTER 7 ITERATIONS
--------------------
SPIN SZ = 0.500
S-SQUARED = 0.750
--------------------
------------
EIGENVECTORS
------------
1 2
3
-1.5563
-0.7970 -0.3007
AG BU
AG
1 H 1 S 0.723161 0.000000
1.124653
2 H 2 S 0.241262 0.753737 -0.860226
3 H 3 S 0.241262 -0.753737 -0.860226
MO1
MO2
MO3



...... END OF ROHF CALCULATION ......
CPU TIME: STEP = 0.11 , TOTAL = 0.8 SECONDS ( 0.0 MIN)
WALL CLOCK TIME: STEP = 0.11 , TOTAL = 0.8 SECONDS ( 0.0 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -1.5562900583
TWO ELECTRON ENERGY = 0.0000000000
NUCLEAR REPULSION ENERGY = 1.3229431231
------------------
TOTAL ENERGY = -0.2333469352
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.0000000000
NUCLEUS-ELECTRON POTENTIAL ENERGY = -2.1242824452
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 1.3229431231
------------------
TOTAL POTENTIAL ENERGY = -0.8013393221
TOTAL KINETIC ENERGY = 0.5679923869
VIRIAL RATIO (V/T) = 1.4108275756
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -1.5562900583
BARE H ENERGY= -1.5562900583
ELECTRONIC ENERGY = -1.5562900583
KINETIC ENERGY= 0.5679923869
N-N REPULSION= 1.3229431231
TOTAL ENERGY= -0.2333469352
SIGMA PART(1+2)= -1.5562900583
(K,V1,2)= 0.5679923869 -2.1242824452 0.0000000000
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= -0.2333469352 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
1.000000
1 0.696294
2 0.151853
3 0.151853
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H 0.696294 0.636499
2 H 0.151853 0.181750
3 H 0.151853 0.181750
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.69629 0.63650
2 H 2 S 0.15185 0.18175
3 H 3 S 0.15185 0.18175
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 0.5229620
2 0.0866661 0.0582075
3 0.0866661 0.0069793 0.0582075
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE
LOW.POP. CHARGE
1 H 0.696294 0.303706
0.636499 0.363501
2 H 0.151853 0.848147
0.181750 0.818250
3 H 0.151853 0.848147
0.181750 0.818250

-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.211 1 3 1.000 0.211
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.908 0.423 0.485
2 H 0.281 0.258 0.023
3 H 0.281 0.258 0.023
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H 1.0 0.3197576
2 H 1.0 0.0540413
3 H 1.0 0.0540413
H3+, Singulett
Input
!
! H3 plus
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=1 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=5000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H3 plus
CNH 2
H 1.0 0.000 0.000 0.000
H 1.0 0.000 1.000 0.000
$END
Output
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
H 1.0 0.0000000000 0.0000000000 0.0000000000
H 1.0 0.0000000000 -1.8897259877 0.0000000000
H 1.0 0.0000000000 1.8897259877 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
H H H
1 H 0.0000000 1.0000000 * 1.0000000 *
2 H 1.0000000 * 0.0000000 2.0000000 *
3 H 1.0000000 * 2.0000000 * 0.0000000
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 1.3229431231
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=F
DENSITY CONV= 1.00E-05
MEMORY REQUIRED FOR RHF STEP= 7602 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0 -1.189979188 -1.189979188 0.121209257 0.000000000
2 1 0 -1.195764362 -0.005785174 0.017147463 0.000000000
3 2 0 -1.195870588 -0.000106226 0.002347024 0.000000000
4 3 0 -1.195872556 -0.000001968 0.000319930 0.000000000
5 4 0 -1.195872592 -0.000000037 0.000043587 0.000000000
6 5 0 -1.195872593 -0.000000001 0.000005938 0.000000000
7 6 0 -1.195872593 0.000000000 0.000000809 0.000000000
-----------------
DENSITY CONVERGED
-----------------
TIME TO FORM FOCK OPERATORS= 0.0 SECONDS ( 0.0 SEC/ITER)
TIME TO SOLVE SCF EQUATIONS= 0.0 SECONDS ( 0.0 SEC/ITER)
FINAL ENERGY IS -1.1958725929 AFTER 7 ITERATIONS
------------
EIGENVECTORS
------------
1 2 3
-0.9742 -0.3372 0.2628
AG BU AG
1 H 1 S 0.583362 0.000000 1.203119
2 H 2 S 0.342482 0.753737 -0.825168
3 H 3 S 0.342482 -0.753737 -0.825168
...... END OF RHF CALCULATION ......
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -3.0892028447
TWO ELECTRON ENERGY = 0.5703871288
NUCLEAR REPULSION ENERGY = 1.3229431231
------------------
TOTAL ENERGY = -1.1958725929
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.5703871288
NUCLEUS-ELECTRON POTENTIAL ENERGY = -4.1459689230
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 1.3229431231
------------------
TOTAL POTENTIAL ENERGY = -2.2526386712
TOTAL KINETIC ENERGY = 1.0567660783
VIRIAL RATIO (V/T) = 2.1316341596
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -1.9484286989
BARE H ENERGY= -3.0892028447
ELECTRONIC ENERGY = -2.5188157718
KINETIC ENERGY= 1.0567660783
N-N REPULSION= 1.3229431231
TOTAL ENERGY= -1.1958726487
SIGMA PART(1+2)= -2.5188157718
(K,V1,2)= 1.0567660783 -4.1459689230 0.5703870729
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= -1.1958726487 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2.000000
1 1.077596
2 0.461202
3 0.461202
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 1.07760 1.03656
2 H 2 S 0.46120 0.48172
3 H 3 S 0.46120 0.48172
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 0.6806230
2 0.1984864 0.2345878
3 0.1984864 0.0281278 0.2345878
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE
LOW.POP. CHARGE
1 H 1.077596 -0.077596 1.036561
-0.036561
2 H 0.461202 0.538798
0.481720 0.518280
3 H 0.461202 0.538798
0.481720 0.518280

-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.497 1 3 1.000 0.497 2 3 2.000 0.213
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.994 0.994 0.000
2 H 0.710 0.710 0.000
3 H 0.710 0.710 0.000
H3+, Triplett
Input
!
! H3 1plus, Triplett
!
$CONTRL SCFTYP=ROHF MULT=3 ICHARG=1 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=5000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H3 1plus, Triplett
CNH 2
H 1.0 0.000 0.000 0.000
H 1.0 0.000 1.000 0.000
$END
Output
H3 1plus, Triplett
THE POINT GROUP OF THE MOLECULE IS CNH
THE ORDER OF THE PRINCIPAL AXIS IS 2
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
H 1.0 0.0000000000 0.0000000000 0.0000000000
H 1.0 0.0000000000 -1.8897259877 0.0000000000
H 1.0 0.0000000000 1.8897259877 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
H H H
1 H 0.0000000 1.0000000 * 1.0000000 *
2 H 1.0000000 * 0.0000000 2.0000000 *
3 H 1.0000000 * 2.0000000 * 0.0000000
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 1.3229431231
MAXIT = 30 NPUNCH= 2 MULT= 3
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
MEMORY REQUIRED FOR UHF/ROHF STEP= 8126 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 -1.042077297 -1.042077297 0.037438296 0.000000000
2 1 -1.043446017 -0.001368720 0.013621524 0.000000000
3 2 -1.043615352 -0.000169335 0.004845675 0.000000000
4 3 -1.043636300 -0.000020948 0.001711033 0.000000000
5 0 -1.043638892 -0.000002591 0.000929753 0.000000000
6 1 -1.043639258 -0.000000366 0.000000013 0.000000000
7 2 -1.043639258 0.000000000 0.000000004 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -1.0436392575 AFTER 7 ITERATIONS
--------------------
SPIN SZ = 1.000
S-SQUARED = 2.000
--------------------
------------
EIGENVECTORS
------------
1 2 3
-1.2063 -0.8237 0.0436
AG BU AG
1 H 1 S 0.572993 0.000000 1.208092
2 H 2 S 0.349566 0.753737 -0.822192
3 H 3 S 0.349566 -0.753737 -0.822192
...... END OF ROHF CALCULATION ......
CPU TIME: STEP = 0.00 , TOTAL = 0.1 SECONDS ( 0.0 MIN)
WALL CLOCK TIME: STEP = 0.00 , TOTAL = 0.1 SECONDS ( 0.0 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -2.7031684789
TWO ELECTRON ENERGY = 0.3365860982
NUCLEAR REPULSION ENERGY = 1.3229431231
------------------
TOTAL ENERGY = -1.0436392575
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.3365860982
NUCLEUS-ELECTRON POTENTIAL ENERGY = -4.1163985968
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 1.3229431231
------------------
TOTAL POTENTIAL ENERGY = -2.4568693754
TOTAL KINETIC ENERGY = 1.4132301179
VIRIAL RATIO (V/T) = 1.7384779338
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -2.0299962831
BARE H ENERGY= -2.7031684789
ELECTRONIC ENERGY = -2.3665823810
KINETIC ENERGY= 1.4132301179
N-N REPULSION= 1.3229431231
TOTAL ENERGY= -1.0436392579
SIGMA PART(1+2)= -2.3665823810
(K,V1,2)= 1.4132301179 -4.1163985968 0.3365860979
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= -1.0436392579 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2
1.000000 1.000000
1 0.527312 0.000000
2 0.236344 0.500000
3 0.236344 0.500000
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H 0.527312 0.509683
2 H 0.736344 0.745159
3 H 0.736344 0.745159
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.52731 0.50968
2 H 2 S 0.73634 0.74516
3 H 3 S 0.73634 0.74516
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3
1 0.3283208
2 0.0994956 0.6903160
3 0.0994956 -0.0534676 0.6903160
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE
LOW.POP. CHARGE
1 H 0.527312 0.472688
0.509683 0.490317
2 H 0.736344 0.263656
0.745159 0.254841
3 H 0.736344 0.263656
0.745159 0.254841

-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.249 1 3 1.000 0.249 2 3 2.000 0.139
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.777 0.499 0.278
2 H 0.930 0.388 0.542
3 H 0.930 0.388 0.542
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H 1.0 0.2225611
2 H 1.0 0.3892425
3 H 1.0 0.3892425
H53+, 31G, Triplett
Input
!
! H5 3plus, CI, Triplett
!
$CONTRL SCFTYP=ROHF MULT=3 ICHARG=3 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=5000000 $END
$BASIS GBASIS=N31 NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H5 3plus, CI, Triplett
CNH 2
H 1.0 0.000 0.000 0.000
H 1.0 0.000 1.000 0.000
H 1.0 0.000 2.000 0.000
$END
Output
H5 3plus, CI, Triplett
THE POINT GROUP OF THE MOLECULE IS CNH
THE ORDER OF THE PRINCIPAL AXIS IS 2
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
H 1.0 0.0000000000 0.0000000000 0.0000000000
H 1.0 0.0000000000 -1.8897259877 0.0000000000
H 1.0 0.0000000000 1.8897259877 0.0000000000
H 1.0 0.0000000000 -3.7794519754 0.0000000000
H 1.0 0.0000000000 3.7794519754 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
H H H H
1 H 0.0000000 1.0000000 * 1.0000000 * 2.0000000 *
2 H 1.0000000 * 0.0000000 2.0000000 * 1.0000000 *
3 H 1.0000000 * 2.0000000 * 0.0000000 3.0000000 *
4 H 2.0000000 * 1.0000000 * 3.0000000 * 0.0000000
5 H 2.0000000 * 3.0000000 * 1.0000000 * 4.0000000
H
1 H 2.0000000 *
2 H 3.0000000 *
3 H 1.0000000 *
4 H 4.0000000
5 H 0.0000000
* ... LESS THAN 3.000
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 3.3955540160
MAXIT = 30 NPUNCH= 2 MULT= 3
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=T
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
SOSCF WILL OPTIMIZE 16 ORBITAL ROTATION ANGLES. SOGTOL= 2.500E-01
MEMORY REQUIRED FOR UHF/ROHF STEP= 9039 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE ORB. GRAD
1 0 -0.269682230 -0.269682230 0.074242758 0.000000000
---------------START SECOND ORDER SCF---------------
2 1 -0.339313280 -0.069631050 0.028597703 0.059613420
3 2 -0.347047636 -0.007734356 0.016457736 0.020764867
4 3 -0.348161298 -0.001113663 0.001230885 0.001268524
5 4 -0.348166907 -0.000005608 0.000561427 0.000510838
6 5 -0.348167631 -0.000000724 0.000036548 0.000051741
7 6 -0.348167636 -0.000000005 0.000009253 0.000015507
8 7 -0.348167636 0.000000000 0.000000621 0.000000474
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -0.3481676362 AFTER 8 ITERATIONS
--------------------
SPIN SZ = 1.000
S-SQUARED = 2.000
--------------------
------------
EIGENVECTORS
------------
1 2 3 4 5
-1.8453 -1.6159 -1.0121 -0.7061 -0.4388
AG BU AG BU AG
1 H 1 S 0.336574 0.000000 0.364970 0.000000 0.335467
2 H 1 S 0.055851 0.000000 0.991379 0.000000 2.541103
3 H 2 S 0.314332 0.379563 -0.069687 -0.258003 -0.327507
4 H 2 S 0.206503 0.359599 -0.510669 -0.689589 -2.137306
5 H 3 S 0.314332 -0.379563 -0.069687 0.258003 -0.327507
6 H 3 S 0.206503 -0.359599 -0.510669 0.689589 -2.137306
7 H 4 S 0.106327 0.190400 -0.304078 0.318766 0.112249
8 H 4 S -0.019058 -0.011156 -0.149541 0.761935 1.236702
9 H 5 S 0.106327 -0.190400 -0.304078 -0.318766 0.112249
10 H 5 S -0.019058 0.011156 -0.149541 -0.761935 1.236702
6 7 8 9 10
-0.1114 0.0384 0.1538 0.2631 0.4195
AG BU AG BU AG
1 H 1 S 0.763498 0.000000 -0.003468 0.000000 -1.122630
2 H 1 S -0.342979 0.000000 0.581704 0.000000 4.920478
3 H 2 S 0.519262 0.446407 0.457796 -0.811574 0.584040
4 H 2 S -0.721373 -0.576436 -1.111025 1.758310 -3.479319
5 H 3 S 0.519262 -0.446407 0.457796 0.811574 0.584040
6 H 3 S -0.721373 0.576436 -1.111025 -1.758310 -3.479319
7 H 4 S 0.301052 0.726772 -0.810056 0.495067 0.269135
8 H 4 S 0.015139 -0.552984 1.221271 -1.239334 0.915163
9 H 5 S 0.301052 -0.726772 -0.810056 -0.495067 0.269135
10 H 5 S 0.015139 0.552984 1.221271 1.239334 0.915163
MO6


MO7


MO8
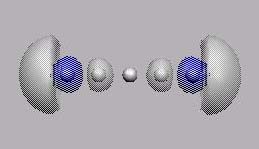

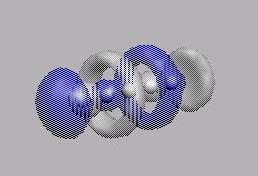
MO10


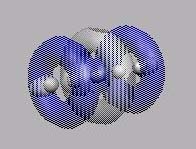
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -4.0263324076
TWO ELECTRON ENERGY = 0.2826107555
NUCLEAR REPULSION ENERGY = 3.3955540160
------------------
TOTAL ENERGY = -0.3481676362
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.2826107555
NUCLEUS-ELECTRON POTENTIAL ENERGY = -5.3947004291
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 3.3955540160
------------------
TOTAL POTENTIAL ENERGY = -1.7165356577
TOTAL KINETIC ENERGY = 1.3683680215
VIRIAL RATIO (V/T) = 1.2544400561
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -3.4611109269
BARE H ENERGY= -4.0263324076
ELECTRONIC ENERGY = -3.7437216673
KINETIC ENERGY= 1.3683680215
N-N REPULSION= 3.3955540160
TOTAL ENERGY= -0.3481676513
SIGMA PART(1+2)= -3.7437216673
(K,V1,2)= 1.3683680215 -5.3947004291 0.2826107404
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= -0.3481676513 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2
1.000000 1.000000
1 0.283463 0.000000
2 0.337448 0.425009
3 0.337448 0.425009
4 0.020821 0.074991
5 0.020821 0.074991
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H 0.283463 0.357861
2 H 0.762457 0.646725
3 H 0.762457 0.646725
4 H 0.095812 0.174344
5 H 0.095812 0.174344
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.23569 0.19342
2 H 1 S 0.04777 0.16445
3 H 2 S 0.42447 0.38611
4 H 2 S 0.33799 0.26062
5 H 3 S 0.42447 0.38611
6 H 3 S 0.33799 0.26062
7 H 4 S 0.10922 0.08992
8 H 4 S -0.01341 0.08442
9 H 5 S 0.10922 0.08992
10 H 5 S -0.01341 0.08442
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 0.1411505
2 0.0712905 0.6799889
3 0.0712905 -0.0421853 0.6799889
4 -0.0001341 0.0538331 -0.0004704 0.0425807
5 -0.0001341 -0.0004704 0.0538331 0.0000025 0.0425807
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H 0.283463 0.716537 0.357861 0.642139
2 H 0.762457 0.237543 0.646725 0.353275
3 H 0.762457 0.237543 0.646725 0.353275
4 H 0.095812 0.904188 0.174344 0.825656
5 H 0.095812 0.904188 0.174344 0.825656
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.191 1 3 1.000 0.191 2 4 1.000 0.141
3 5 1.000 0.141
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.487 0.406 0.080
2 H 0.934 0.344 0.590
3 H 0.934 0.344 0.590
4 H 0.182 0.173 0.009
5 H 0.182 0.173 0.009
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H 1.0 0.1704417
2 H 1.0 0.3379132
3 H 1.0 0.3379132
4 H 1.0 0.0764442
5 H 1.0 0.0764442