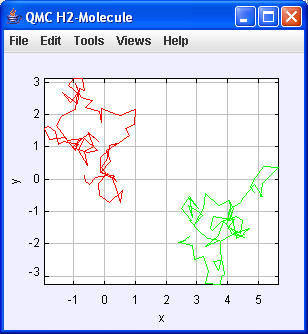
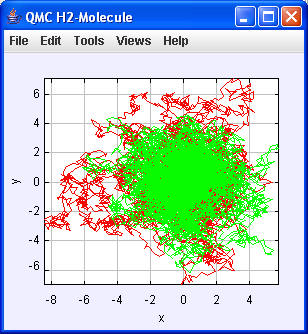
QMC_H2
| N = 100
|
N = 10000
|
Run I/O:
----jGRASP exec: java qmcH2.MonteCarlo 100
Integrating...
r1 in Stabilising: -0.8105120797046247,0.06259845647383633,0.5586589974492433
x1 mit r1.getComponent(0): -0.8105120797046247
pot. Energy: -0.9274765038394915 Var: -0.012578247210878135
Energy (Virial Theorem): -0.46373825191974577
----jGRASP: operation complete.
----jGRASP exec: java qmcH2.MonteCarlo 10000
Integrating...
r1 in Stabilising: 0.8317117155546266,-2.298805229125598,2.0149717993222036
x1 mit r1.getComponent(0): 0.8317117155546266
pot. Energy: -1.1783991953195483 Var: -2.3591177085201525E-4
Energy (Virial Theorem): -0.5891995976597741
MonteCarlo.java
package qmcH2;
import java.io.*;
import JSci.maths.*;
import JSci.maths.statistics.*;
import JSci.io.*;
import java.awt.Color;
import org.opensourcephysics.frames.PlotFrame;
import org.opensourcephysics.display.*;
/**
* Monte Carlo calculation of Helium ground state energy.
* @author Mark Hale
* @version 1.0
*/
/**
* Monte Carlo calculation of H2-molecule ground state energy.
* @author Guenter Haucke
* @version 1.0
*/
public class MonteCarlo implements Mapping
{
private int N;
private double potE[];
private double stepW;
// Walker step width
private DoubleVector r1; // Vector
of electron1
private DoubleVector r2;
private DoubleVector nucR1; //
Vector of nucleus1
private DoubleVector nucR2;
private double R;
PlotFrame frame = new PlotFrame("x", "y", "QMC H2-Molecule");
/**
* Instantiate class.
*/
public static void main(String arg[])
{
if(arg.length==1)
{
int n=Integer.valueOf(arg[0]).intValue();
new MonteCarlo(n);
}
else
{
System.out.println("Need to specify command line arguments:");
System.out.println("<number of iterations>");
}
}
/**
* Constructor.
* @param n number of iterations
*/
public MonteCarlo(int n)
{
N=n;
R = 1.4; // distance between the
two nuclei in atomic units; experimental value: 1.40
r1=new Double3Vector(Math.random(),Math.random(),Math.random());
r2=new Double3Vector(-Math.random(),-Math.random(),-Math.random());
nucR1=new Double3Vector(R/2,0.0,0.0);
// nucleus1 is set to coordinates x=R/2,
y=0, z=0
nucR2=new Double3Vector(-R/2,0.0,0.0);
// nucleus2 is set to coordinates
x=-R/2, y=0, z=0
//energy=new double[N];
potE = new double[N];
frame.setConnected(0, true);
frame.setMarkerShape(0, Dataset.NO_MARKER);
frame.setConnected(1, true);
frame.setMarkerShape(1, Dataset.NO_MARKER);
frame.setVisible(true);
// frame.repaint();
compute();
saveResults();
frame.setDefaultCloseOperation(javax.swing.JFrame.EXIT_ON_CLOSE);
frame.setXPointsLinked(true); // default is true
frame.setXYColumnNames(0,"x1","y1");
//frame.setXYColumnNames(1,"x2","y2");
frame.setRowNumberVisible(true);
}
/**
* Compute the local potential energy.
*/
private void compute()
{
DoubleVector tmpr1,tmpr2;
double prob,tmpprob;
// Stabilising
for(int i=0;i<1000;i++)
{
tmpr1=r1.mapComponents(this);
tmpr2=r2.mapComponents(this);
tmpprob=trialWF(tmpr1, tmpr2);
tmpprob*=tmpprob;
prob=trialWF(r1, r2);
prob*=prob;
if(tmpprob/prob>Math.random())
{
r1=tmpr1;
r2=tmpr2;
}
}
System.out.println("Integrating...");
System.out.println("r1 in Stabilising: " + r1);
System.out.println("x1 mit r1.getComponent(0): " + r1.getComponent(0));
for(int i=0;i<N;i++)
{
tmpr1=r1.mapComponents(this);
tmpr2=r2.mapComponents(this);
tmpprob=trialWF(tmpr1, tmpr2);
tmpprob*=tmpprob;
prob=trialWF(r1, r2);
prob*=prob;
if(tmpprob/prob>Math.random())
{
r1=tmpr1;
r2=tmpr2;
}
//energy[i]=localEnergy(r1,r2);
potE[i]=localpotE(r1, r2);
frame.append(0, r1.getComponent(0), r1.getComponent(1));
frame.append(1, r2.getComponent(0), r2.getComponent(1));
frame.setVisible(true);
frame.repaint();
}
}
/**
* Trial wavefunction.
* @param r1 position vector of electron 1
* @param r2 position vector of electron 2
*/
private double trialWF(DoubleVector r1, DoubleVector r2)
{
double modR1=r1.norm();
double modR2=r2.norm();
double modR12=r1.subtract(r2).norm();
//return Math.exp(-1*modR1)*Math.exp(-1*modR2)*Math.exp(modR12/2);
// wavefunction including el-el
repulsion
return Math.exp(-1*modR1)*Math.exp(-1*modR2);
}
private double localpotE(DoubleVector r1, DoubleVector r2)
{
DoubleVector r12=r2.subtract(r1);
// interelectronic distance
DoubleVector nucR12=nucR2.subtract(nucR1);
// distance between the two nuclei
DoubleVector r1R1=r1.subtract(nucR1);
// distance between el1 and nuc1
DoubleVector r2R2=r2.subtract(nucR2);
// distance between el2 and nuc2
DoubleVector r1R2=r1.subtract(nucR2);
// distance between el1 and nuc2
DoubleVector r2R1=r2.subtract(nucR1);
// distance between el2 and nuc1
double termr1R1=-1/(r1R1.norm());
double termr2R2=-1/(r2R2.norm());
double termr1R2=-1/(r1R2.norm());
double termr2R1=-1/(r2R1.norm());
return termr1R1 + termr2R2 + + termr1R2 + termr2R1 + 1/r12.norm() +
1/nucR12.norm();
}
/**
* Update electron co-ordinates.
*/
public double map(double x)
{
stepW = 0.5;
return x+(2*Math.random()-1.0)*stepW;
}
/**
* Log results to disk.
*/
private void saveResults()
{
File file=new File("results.dat");
char buf[]=null;
NormalDistribution normpot = new NormalDistribution(potE);
double data[][]=new double[1][3];
double meanpot = normpot.getMean();
double varpot = normpot.getVariance();
System.out.println("pot. Energy: "+ meanpot + " Var: " + varpot);
System.out.println("Energy (Virial Theorem): " + meanpot/2);
data[0][0]=N;
data[0][1]=meanpot;
data[0][2]=varpot;
// Read in existing data
try
{
FileReader in=new FileReader(file);
buf=new char[(int)file.length()];
in.read(buf);
in.close();
}
catch(Exception e)
{
System.out.println("No previous data - new file.");
}
// Save all to file
try
{
TextWriter out=new TextWriter(file,',');
if(buf!=null)
out.write(buf);
out.write(data);
out.close();
}
catch(Exception e)
{
System.out.println("Failed to save.");
}
}
}