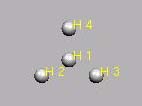
H43+ (1 Elektron)
Input
!
! H4-3+-Molekuel, verzweigt
!
$CONTRL SCFTYP=ROHF MULT=2 ICHARG=+3 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=1000 MEMORY=5000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H4-3+-Molekuel verzweigt
CNH 3
H 1.0 0.000 0.000 0.000
H 1.0 0.000 1.000 0.000
$END
Output
RUN TITLE
---------
H4-3+-Molekuel verzweigt
THE POINT GROUP OF THE MOLECULE IS CNH
THE ORDER OF THE PRINCIPAL AXIS IS 3
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X
Y
Z
H 1.0 0.0000000000
0.0000000000 0.0000000000
H 1.0 -1.6365507116 -0.9448629939
0.0000000000
H 1.0 1.6365507116 -0.9448629939
0.0000000000
H 1.0 0.0000000000
1.8897259877 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
H
H H
H
1 H 0.0000000 1.0000000 *
1.0000000 * 1.0000000 *
2 H 1.0000000 * 0.0000000
1.7320508 * 1.7320508 *
3 H 1.0000000 * 1.7320508 * 0.0000000
1.7320508 *
4 H 1.0000000 * 1.7320508 * 1.7320508
* 0.0000000
* ... LESS THAN 3.000
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 2.5040936296
MAXIT = 30 NPUNCH= 2 MULT= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
MEMORY REQUIRED FOR UHF/ROHF STEP= 8197 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0.521807221 0.521807221 0.268589904 0.000000000
2 1 0.467188771 -0.054618450 0.097272218 0.000000000
3 2 0.462052854 -0.005135917 0.031031336 0.000000000
4 3 0.461568069 -0.000484785 0.009636169 0.000000000
5 0 0.461522295 -0.000045774 0.004287956 0.000000000
6 1 0.461517523 -0.000004773 0.000000975 0.000000000
7 2 0.461517523 0.000000000 0.000000300 0.000000000
-----------------
DENSITY CONVERGED
-----------------
------------
EIGENVECTORS
Singulett
Triplett
------------
1 2
3 4
-2.0426
-1.0690 -1.0690 -0.5941
A' E'
E' A'
1 H 1 S 0.786204 0.000000
0.000000 1.252110
2 H 2 S 0.132367 -0.451446 -0.781927
-0.718677
3 H 3 S 0.132367 -0.451446 0.781927
-0.718677
4 H 4 S 0.132367 0.902892
0.000000 -0.718677
MO1
MO2
MO3



MO4

...... END OF ROHF CALCULATION ......
CPU TIME: STEP = 0.00 , TOTAL = 0.2 SECONDS ( 0.0 MIN)
WALL CLOCK TIME: STEP = 0.00 , TOTAL = 0.2 SECONDS ( 0.0 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -2.0425761070
TWO ELECTRON ENERGY = 0.0000000000
NUCLEAR REPULSION ENERGY = 2.5040936296
------------------
TOTAL ENERGY = 0.4615175226
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.0000000000
NUCLEUS-ELECTRON POTENTIAL ENERGY = -2.6271688682
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 2.5040936296
------------------
TOTAL POTENTIAL ENERGY = -0.1230752386
TOTAL KINETIC ENERGY = 0.5845927612
VIRIAL RATIO (V/T) = 0.2105315816
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -2.0425761070
BARE H ENERGY= -2.0425761070
ELECTRONIC ENERGY = -2.0425761070
KINETIC ENERGY= 0.5845927612
N-N REPULSION= 2.5040936296
TOTAL ENERGY= 0.4615175226
SIGMA PART(1+2)= -2.0425761070
(K,V1,2)= 0.5845927612 -2.6271688682 0.0000000000
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= 0.4615175226 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
1.000000
1 0.773199
2 0.075600
3 0.075600
4 0.075600
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H 0.773199 0.652739
2 H 0.075600 0.115754
3 H 0.075600 0.115754
4 H 0.075600 0.115754
----- POPULATIONS IN EACH AO -----
MULLIKEN
LOWDIN
1 H 1 S 0.77320 0.65274
2 H 2 S 0.07560 0.11575
3 H 3 S 0.07560 0.11575
4 H 4 S 0.07560 0.11575
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4
1 0.6181170
2 0.0516940 0.0175210
3 0.0516940 0.0031927 0.0175210
4 0.0516940 0.0031927 0.0031927
0.0175210
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE
LOW.POP. CHARGE
1 H 0.773199 0.226801
0.652739 0.347261
2 H 0.075600 0.924400
0.115754 0.884246
3 H 0.075600 0.924400
0.115754 0.884246
4 H 0.075600 0.924400
0.115754 0.884246
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND
BOND
BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.117 1 3 1.000
0.117 1 4 1.000 0.117
TOTAL BONDED
FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.949 0.351
0.598
2 H 0.145 0.140
0.006
3 H 0.145 0.140
0.006
4 H 0.145 0.140
0.006
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H 1.0 0.3662956
2 H 1.0 0.0253816
3 H 1.0 0.0253816
4 H 1.0 0.0253816
Input
!
! H4 2plus, Singulett
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=2 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=1000 MEMORY=5000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H2 2plus, Singulett
DNH 2
H 1.0 0.000 0.500 0.000
H 1.0 0.000 1.500 0.000
$END
Output
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 2.5040936296
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=F
DENSITY CONV= 1.00E-05
MEMORY REQUIRED FOR RHF STEP= 7665 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0 -0.934833604 -0.934833604 0.292668609 0.000000000
2 1 0 -0.967102525 -0.032268922 0.043867736 0.000000000
3 2 0 -0.967681065 -0.000578539 0.005943642 0.000000000
4 3 0 -0.967691405 -0.000010340 0.000796041 0.000000000
5 4 0 -0.967691590 -0.000000185 0.000106454 0.000000000
6 5 0 -0.967691593 -0.000000003 0.000014233 0.000000000
7 6 0 -0.967691593 0.000000000 0.000001903 0.000000000
8 7 0 -0.967691593 0.000000000 0.000000254 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -0.9676915929 AFTER 8 ITERATIONS
------------
EIGENVECTORS
Triplett
Ein-Elektron
------------
1 2
3 4
-1.4464
-0.6350 -0.6350 -0.0377
A' E'
E' A'
1 H 1 S 0.615628 0.000000
0.000000 1.344210
2 H 2 S 0.225257 -0.451446 -0.781927
-0.695181
3 H 3 S 0.225257 -0.451446 0.781927
-0.695181
4 H 4 S 0.225257 0.902892
0.000000 -0.695181
...... END OF RHF CALCULATION ......
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -4.0508002646
TWO ELECTRON ENERGY = 0.5790150420
NUCLEAR REPULSION ENERGY = 2.5040936296
------------------
TOTAL ENERGY = -0.9676915929
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.5790150420
NUCLEUS-ELECTRON POTENTIAL ENERGY = -5.0497530798
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 2.5040936296
------------------
TOTAL POTENTIAL ENERGY = -1.9666444082
TOTAL KINETIC ENERGY = 0.9989528153
VIRIAL RATIO (V/T) = 1.9687060071
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -2.8927702205
BARE H ENERGY= -4.0508002646
ELECTRONIC ENERGY = -3.4717852425
KINETIC ENERGY= 0.9989528153
N-N REPULSION= 2.5040936296
TOTAL ENERGY= -0.9676916129
SIGMA PART(1+2)= -3.4717852425
(K,V1,2)= 0.9989528153 -5.0497530798 0.5790150220
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= -0.9676916129 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2.000000
1 1.171301
2 0.276233
3 0.276233
4 0.276233

----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 1.17130 1.04800
2 H 2 S 0.27623 0.31733
3 H 3 S 0.27623 0.31733
4 H 4 S 0.27623 0.31733
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4
1 0.7579951
2 0.1377686 0.1014810
3 0.1377686 0.0184917 0.1014810
4 0.1377686 0.0184917 0.0184917 0.1014810
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H 1.171301 -0.171301 1.048002 -0.048002
2 H 0.276233 0.723767 0.317333 0.682667
3 H 0.276233 0.723767 0.317333 0.682667
4 H 0.276233 0.723767 0.317333 0.682667
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.324 1 3 1.000 0.324 1 4 1.000 0.324
2 3 1.732 0.076 2 4 1.732 0.076 3 4 1.732 0.076
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.971 0.971 0.000
2 H 0.476 0.476 0.000
3 H 0.476 0.476 0.000
4 H 0.476 0.476 0.000
Triplett
Input
!
! H4-2+-Molekuel, verzweigt, Triplett
!
$CONTRL SCFTYP=ROHF MULT=3 ICHARG=+2 RUNTYP=ENERGY COORD=UNIQUE $END
$SYSTEM TIMLIM=1000 MEMORY=5000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H4-2+-Molekuel verzweigt, Triplett
CNH 3
H 1.0 0.000 0.000 0.000
H 1.0 0.000 1.000 0.000
$END
Output
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 2.5040936296
MAXIT = 30 NPUNCH= 2 MULT= 3
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
MEMORY REQUIRED FOR UHF/ROHF STEP= 8197 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 -0.375854694 -0.375854694 0.022728923 0.000000000
2 1 -0.372936484 0.002918210 0.006336234 0.000000000
3 2 -0.373731533 -0.000795049 0.001965214 0.000000000
4 3 -0.373966211 -0.000234678 0.000605679 0.000000000
5 4 -0.374037424 -0.000071213 0.000186312 0.000000000
6 5 -0.374059225 -0.000021801 0.000057277 0.000000000
7 6 -0.374065917 -0.000006692 0.000017605 0.000000000
8 7 -0.374067973 -0.000002056 0.000005411 0.000000000
9 8 -0.374068605 -0.000000632 0.000001663 0.000000000
10 9 -0.374068799 -0.000000194 0.000000511 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS -0.3740687990 AFTER 10 ITERATIONS
--------------------
SPIN SZ = 1.000
S-SQUARED = 2.000
--------------------
------------
EIGENVECTORS
Singulett
Ein-Elektron
------------
1 2
3 4
-1.3484
-0.9686 -0.9686 0.0999
A' E'
E' A'
1 H 1 S 0.456977 0.000000
0.000000 1.406083
2 H 2 S 0.303700 -0.451446 -0.781927
-0.664668
3 H 3 S 0.303700 -0.451446 0.781927
-0.664668
4 H 4 S 0.303700 0.902892
0.000000 -0.664668
...... END OF ROHF CALCULATION ......
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -3.4394007896
TWO ELECTRON ENERGY = 0.5612383610
NUCLEAR REPULSION ENERGY = 2.5040936296
------------------
TOTAL ENERGY = -0.3740687990
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.5612383610
NUCLEUS-ELECTRON POTENTIAL ENERGY = -4.8491465192
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 2.5040936296
------------------
TOTAL POTENTIAL ENERGY = -1.7838145286
TOTAL KINETIC ENERGY = 1.4097457296
VIRIAL RATIO (V/T) = 1.2653448711
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -2.3169242564
BARE H ENERGY= -3.4394007896
ELECTRONIC ENERGY = -2.8781625230
KINETIC ENERGY= 1.4097457296
N-N REPULSION= 2.5040936296
TOTAL ENERGY= -0.3740688934
SIGMA PART(1+2)= -2.8781625230
(K,V1,2)= 1.4097457296 -4.8491465192 0.5612382666
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= -0.3740688934 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2
1.000000 1.000000
1 0.415644 0.000000
2 0.194785 0.166667
3 0.194785 0.166667
4 0.194785 0.666667

ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H 0.415644 0.409273
2 H 0.361452 0.363576
3 H 0.361452 0.363576
4 H 0.861452 0.863576
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.41564 0.40927
2 H 2 S 0.36145 0.36358
3 H 3 S 0.36145 0.36358
4 H 4 S 0.86145 0.86358
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4
1 0.2088275
2 0.0689387 0.2960369
3 0.0689387 0.0539434 0.2960369
4 0.0689387 -0.0574668 -0.0574668 0.9074471
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H 0.415644 0.584356 0.409273 0.590727
2 H 0.361452 0.638548 0.363576 0.636424
3 H 0.361452 0.638548 0.363576 0.636424
4 H 0.861452 0.138548 0.863576 0.136424
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.162 1 3 1.000 0.162 1 4 1.000 0.162
2 3 1.732 0.261
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H 0.659 0.486 0.173
2 H 0.592 0.462 0.131
3 H 0.592 0.462 0.131
4 H 0.981 0.239 0.742
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H 1.0 0.1621738
2 H 1.0 0.1752840
3 H 1.0 0.1752840
4 H 1.0 0.4909283
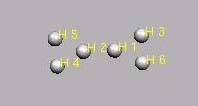
Input, H65+
!
! H6, verzweigt, zwei Dreiringe
!
$CONTRL SCFTYP=ROHF MULT=2 ICHARG=5 RUNTYP=ENERGY COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H6, verzweigt, zwei Dreiringe
CNH 2
H1
H2 1 1.0
H3 2 1.0 1 150.00
H4 2 1.0 1 150.00 3 180.00 0
H5 1 1.0 2 150.00 3 180.00 0
H6 1 1.0 2 150.00 3 0.00 0
$END
Output
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 5.5510961775
MAXIT = 30 NPUNCH= 2 MULT= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
MEMORY REQUIRED FOR UHF/ROHF STEP= 8387 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 2.960007659 2.960007659 0.105080250 0.000000000
2 1 2.893711713 -0.066295946 0.038031986 0.000000000
3 2 2.886036213 -0.007675499 0.012983067 0.000000000
4 3 2.885147644 -0.000888570 0.004413679 0.000000000
5 0 2.885044793 -0.000102851 0.002272306 0.000000000
6 1 2.885031330 -0.000013463 0.000001536 0.000000000
7 2 2.885031330 0.000000000 0.000000523 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS 2.8850313299 AFTER 7 ITERATIONS
--------------------
SPIN SZ = 0.500
S-SQUARED = 0.750
--------------------
------------
EIGENVECTORS
------------
1 2
3 4
5
-2.6661
-2.0984 -1.6549 -1.3858
-1.3550
AG AU
AG BU
BG
1 H 1 S 0.516067 0.476732
0.490494 0.000000 0.000000
2 H 2 S 0.516067 -0.476732 0.490494
0.000000 0.000000
3 H 3 S 0.067126 0.265186 -0.488985
0.698306 0.711498
4 H 4 S 0.067126 -0.265186 -0.488985
-0.698306 0.711498
5 H 5 S 0.067126 -0.265186 -0.488985
0.698306 -0.711498
6 H 6 S 0.067126 0.265186 -0.488985
-0.698306 -0.711498
MO1
MO2
MO3


MO4


6
-1.1911
AU
1 H 1 S 1.156198
2 H 2 S -1.156198
3 H 3 S -0.451331
4 H 4 S 0.451331
5 H 5 S 0.451331
6 H 6 S -0.451331
MO6

...... END OF ROHF CALCULATION ......
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -2.6660648476
TWO ELECTRON ENERGY = 0.0000000000
NUCLEAR REPULSION ENERGY = 5.5510961775
------------------
TOTAL ENERGY = 2.8850313299
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.0000000000
NUCLEUS-ELECTRON POTENTIAL ENERGY = -3.1784888687
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 5.5510961775
------------------
TOTAL POTENTIAL ENERGY = 2.3726073089
TOTAL KINETIC ENERGY = 0.5124240210
VIRIAL RATIO (V/T) = -4.6301641054
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -2.6660648476
BARE H ENERGY= -2.6660648476
ELECTRONIC ENERGY = -2.6660648476
KINETIC ENERGY= 0.5124240210
N-N REPULSION= 5.5510961775
TOTAL ENERGY= 2.8850313299
SIGMA PART(1+2)= -2.6660648476
(K,V1,2)= 0.5124240210 -3.1784888687 0.0000000000
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= 2.8850313299 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
1.000000
1 0.442292
2 0.442292
3 0.028854
4 0.028854
5 0.028854
6 0.028854
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H1 0.442292 0.404853
2 H1 0.442292 0.404853
3 H3 0.028854 0.047574
4 H3 0.028854 0.047574
5 H3 0.028854 0.047574
6 H3 0.028854 0.047574
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.44229 0.40485
2 H 2 S 0.44229 0.40485
3 H 3 S 0.02885 0.04757
4 H 4 S 0.02885 0.04757
5 H 5 S 0.02885 0.04757
6 H 6 S 0.02885 0.04757
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 0.2663249
2 0.1322929 0.2663249
3 0.0172077 0.0046295 0.0045060
4 0.0046295 0.0172077 0.0001150 0.0045060
5 0.0046295 0.0172077 0.0001575 0.0022383 0.0045060
6 0.0172077 0.0046295 0.0022383 0.0001575 0.0001150
6
6 0.0045060
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H1 0.442292 0.557708 0.404853 0.595147
2 H1 0.442292 0.557708 0.404853 0.595147
3 H3 0.028854 0.971146 0.047574 0.952426
4 H3 0.028854 0.971146 0.047574 0.952426
5 H3 0.028854 0.971146 0.047574 0.952426
6 H3 0.028854 0.971146 0.047574 0.952426
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.391
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H1 0.689 0.493 0.196
2 H1 0.689 0.493 0.196
3 H3 0.057 0.056 0.001
4 H3 0.057 0.056 0.001
5 H3 0.057 0.056 0.001
6 H3 0.057 0.056 0.001
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H1 1.0 0.1828171
2 H1 1.0 0.1828171
3 H3 1.0 0.0094647
4 H3 1.0 0.0094647
5 H3 1.0 0.0094647
6 H3 1.0 0.0094647
H64+, Singulett
Input
!
! H6, verzweigt, zwei Dreiringe, 4+, Singulett
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=4 RUNTYP=ENERGY COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H6, verzweigt, zwei Dreiringe, 4+, Singulett
CNH 2
H1
H2 1 1.0
H3 2 1.0 1 150.00
H4 2 1.0 1 150.00 3 180.00 0
H5 1 1.0 2 150.00 3 180.00 0
H6 1 1.0 2 150.00 3 0.00 0
$END
Output
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 5.5510961775
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=F
DENSITY CONV= 1.00E-05
MEMORY REQUIRED FOR RHF STEP= 7833 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0 0.812113909 0.812113909 0.145317563 0.000000000
2 1 0 0.757695880 -0.054418029 0.023918093 0.000000000
3 2 0 0.756405789 -0.001290091 0.004067538 0.000000000
4 3 0 0.756369041 -0.000036748 0.000697534 0.000000000
5 0 0 0.756367963 -0.000001078 0.000144644 0.000000000
6 1 0 0.756367930 -0.000000033 0.000000003 0.000000000
7 2 0 0.756367930 0.000000000 0.000000000 0.000000000
-----------------
DENSITY CONVERGED
-----------------
TIME TO FORM FOCK OPERATORS= 0.0 SECONDS ( 0.0 SEC/ITER)
TIME TO SOLVE SCF EQUATIONS= 0.0 SECONDS ( 0.0 SEC/ITER)
FINAL ENERGY IS 0.7563679300 AFTER 7 ITERATIONS
------------
EIGENVECTORS
------------
1 2 3 4 5
-2.1437 -1.6889 -1.2321 -1.0244 -0.9927
AG AU AG BU BG
1 H 1 S 0.436594 0.347434 0.562401 0.000000 0.000000
2 H 2 S 0.436594 -0.347434 0.562401 0.000000 0.000000
3 H 3 S 0.139765 0.312952 -0.473369 0.698306 0.711498
4 H 4 S 0.139765 -0.312952 -0.473369 -0.698306 0.711498
5 H 5 S 0.139765 -0.312952 -0.473369 0.698306 -0.711498
6 H 6 S 0.139765 0.312952 -0.473369 -0.698306 -0.711498
6
-0.6797
AU
1 H 1 S 1.201398
2 H 2 S -1.201398
3 H 3 S -0.419624
4 H 4 S 0.419624
5 H 5 S 0.419624
6 H 6 S -0.419624
...... END OF RHF CALCULATION ......
CPU TIME: STEP = 0.06 , TOTAL = 0.4 SECONDS ( 0.0 MIN)
WALL CLOCK TIME: STEP = 0.06 , TOTAL = 0.4 SECONDS ( 0.0 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -5.3020640908
TWO ELECTRON ENERGY = 0.5073358433
NUCLEAR REPULSION ENERGY = 5.5510961775
------------------
TOTAL ENERGY = 0.7563679300
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.5073358433
NUCLEUS-ELECTRON POTENTIAL ENERGY = -6.2070180286
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 5.5510961775
------------------
TOTAL POTENTIAL ENERGY = -0.1485860078
TOTAL KINETIC ENERGY = 0.9049539378
VIRIAL RATIO (V/T) = 0.1641917910
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -4.2873924044
BARE H ENERGY= -5.3020640908
ELECTRONIC ENERGY = -4.7947282476
KINETIC ENERGY= 0.9049539378
N-N REPULSION= 5.5510961775
TOTAL ENERGY= 0.7563679299
SIGMA PART(1+2)= -4.7947282476
(K,V1,2)= 0.9049539378 -6.2070180286 0.5073358432
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= 0.7563679299 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2.000000
1 0.724461
2 0.724461
3 0.137769
4 0.137769
5 0.137769
6 0.137769

----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.72446 0.67924
2 H 2 S 0.72446 0.67924
3 H 3 S 0.13777 0.16038
4 H 4 S 0.13777 0.16038
5 H 5 S 0.13777 0.16038
6 H 6 S 0.13777 0.16038
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 0.3812284
2 0.1893695 0.3812284
3 0.0606222 0.0163094 0.0390686
4 0.0163094 0.0606222 0.0009973 0.0390686
5 0.0163094 0.0606222 0.0013652 0.0194068 0.0390686
6 0.0606222 0.0163094 0.0194068 0.0013652 0.0009973
6
6 0.0390686
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H1 0.724461 0.275539 0.679242 0.320758
2 H1 0.724461 0.275539 0.679242 0.320758
3 H3 0.137769 0.862231 0.160379 0.839621
4 H3 0.137769 0.862231 0.160379 0.839621
5 H3 0.137769 0.862231 0.160379 0.839621
6 H3 0.137769 0.862231 0.160379 0.839621
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.525 1 3 1.000 0.100 1 4 1.932 0.100
1 5 1.932 0.100 1 6 1.000 0.100 2 3 1.932 0.100
2 4 1.000 0.100 2 5 1.000 0.100 2 6 1.932 0.100
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H1 0.924 0.924 0.000
2 H1 0.924 0.924 0.000
3 H3 0.257 0.257 0.000
4 H3 0.257 0.257 0.000
5 H3 0.257 0.257 0.000
6 H3 0.257 0.257 0.000
H64+, Triplett
Input
!
! H6, verzweigt, zwei Dreiringe, 4+, Triplett
!
$CONTRL SCFTYP=ROHF MULT=3 ICHARG=4 RUNTYP=ENERGY COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H6, verzweigt, zwei Dreiringe, 4+, Triplett
CNH 2
H1
H2 1 1.0
H3 2 1.0 1 150.00
H4 2 1.0 1 150.00 3 180.00 0
H5 1 1.0 2 150.00 3 180.00 0
H6 1 1.0 2 150.00 3 0.00 0
$END
Output
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 5.5510961775
MAXIT = 30 NPUNCH= 2 MULT= 3
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
MEMORY REQUIRED FOR UHF/ROHF STEP= 8387 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0.841167420 0.841167420 0.174057823 0.000000000
2 1 0.781160343 -0.060007077 0.065451315 0.000000000
3 2 0.775257494 -0.005902848 0.020983487 0.000000000
4 3 0.774666016 -0.000591478 0.006538415 0.000000000
5 4 0.774605133 -0.000060884 0.002034373 0.000000000
6 5 0.774598702 -0.000006430 0.000636566 0.000000000
7 0 0.774598008 -0.000000694 0.000294028 0.000000000
8 1 0.774597922 -0.000000086 0.000000447 0.000000000
9 2 0.774597922 0.000000000 0.000000133 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS 0.7745979222 AFTER 9 ITERATIONS
--------------------
SPIN SZ = 1.000
S-SQUARED = 2.000
--------------------
------------
EIGENVECTORS
------------
1 2 3 4 5
-2.3509 -2.1224 -1.3373 -1.0642 -1.0376
AG AU AG BU BG
1 H 1 S 0.445575 0.477053 0.555312 0.000000 0.000000
2 H 2 S 0.445575 -0.477053 0.555312 0.000000 0.000000
3 H 3 S 0.132140 0.265061 -0.475554 0.698306 0.711498
4 H 4 S 0.132140 -0.265061 -0.475554 -0.698306 0.711498
5 H 5 S 0.132140 -0.265061 -0.475554 0.698306 -0.711498
6 H 6 S 0.132140 0.265061 -0.475554 -0.698306 -0.711498
6
-0.8396
AU
1 H 1 S 1.156066
2 H 2 S -1.156066
3 H 3 S -0.451405
4 H 4 S 0.451405
5 H 5 S 0.451405
6 H 6 S -0.451405
...... END OF ROHF CALCULATION ......
CPU TIME: STEP = 0.00 , TOTAL = 0.3 SECONDS ( 0.0 MIN)
WALL CLOCK TIME: STEP = 0.00 , TOTAL = 0.3 SECONDS ( 0.0 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -5.0796115130
TWO ELECTRON ENERGY = 0.3031132577
NUCLEAR REPULSION ENERGY = 5.5510961775
------------------
TOTAL ENERGY = 0.7745979222
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.3031132577
NUCLEUS-ELECTRON POTENTIAL ENERGY = -6.2114642216
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 5.5510961775
------------------
TOTAL POTENTIAL ENERGY = -0.3572547863
TOTAL KINETIC ENERGY = 1.1318527085
VIRIAL RATIO (V/T) = 0.3156371705
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -4.4733850205
BARE H ENERGY= -5.0796115130
ELECTRONIC ENERGY = -4.7764982668
KINETIC ENERGY= 1.1318527085
N-N REPULSION= 5.5510961775
TOTAL ENERGY= 0.7745979108
SIGMA PART(1+2)= -4.7764982668
(K,V1,2)= 1.1318527085 -6.2114642216 0.3031132462
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= 0.7745979108 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2
1.000000 1.000000
1 0.371389 0.206358
2 0.371389 0.206358
3 0.064306 0.146821
4 0.064306 0.146821
5 0.064306 0.146821
6 0.064306 0.146821

ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H1 0.577747 0.520462
2 H1 0.577747 0.520462
3 H3 0.211127 0.239769
4 H3 0.211127 0.239769
5 H3 0.211127 0.239769
6 H3 0.211127 0.239769
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.57775 0.52046
2 H 2 S 0.57775 0.52046
3 H 3 S 0.21113 0.23977
4 H 4 S 0.21113 0.23977
5 H 5 S 0.21113 0.23977
6 H 6 S 0.21113 0.23977
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 0.4261167
2 -0.0144261 0.4261167
3 0.0920581 -0.0090299 0.0877182
4 -0.0090299 0.0920581 -0.0013477 0.0877182
5 -0.0090299 0.0920581 -0.0018449 0.0435727 0.0877182
6 0.0920581 -0.0090299 0.0435727 -0.0018449 -0.0013477
6
6 0.0877182
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H1 0.577747 0.422253 0.520462 0.479538
2 H1 0.577747 0.422253 0.520462 0.479538
3 H3 0.211127 0.788873 0.239769 0.760231
4 H3 0.211127 0.788873 0.239769 0.760231
5 H3 0.211127 0.788873 0.239769 0.760231
6 H3 0.211127 0.788873 0.239769 0.760231
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.000 0.054 1 3 1.000 0.216 1 6 1.000 0.216
2 4 1.000 0.216 2 5 1.000 0.216 3 6 1.000 0.089
4 5 1.000 0.089
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H1 0.822 0.488 0.334
2 H1 0.822 0.488 0.334
3 H3 0.378 0.333 0.045
4 H3 0.378 0.333 0.045
5 H3 0.378 0.333 0.045
6 H3 0.378 0.333 0.045
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H1 1.0 0.2671569
2 H1 1.0 0.2671569
3 H3 1.0 0.0807561
4 H3 1.0 0.0807561
5 H3 1.0 0.0807561
6 H3 1.0 0.0807561
H6, zwei Vierringe

Input, H65+
!
! H6, verzeigt,wie Bicyclobut
!
$CONTRL SCFTYP=ROHF MULT=2 ICHARG=5 RUNTYP=ENERGY COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H6, verzweigt, wie Bicyclobut
CNH 2
H1
H2 1 1.55
H3 1 1.38 2 87.8
H4 2 1.38 1 87.8 3 0.0 0
H5 1 1.38 2 87.8 4 -180.0 0
H6 2 1.38 1 87.8 3 180.0 0
$END
Output
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 4.3718366655
MAXIT = 30 NPUNCH= 2 MULT= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
MEMORY REQUIRED FOR UHF/ROHF STEP= 8387 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 2.187980841 2.187980841 0.087464433 0.000000000
2 1 2.152644494 -0.035336347 0.035198703 0.000000000
3 2 2.147402651 -0.005241842 0.013542482 0.000000000
4 3 2.146630784 -0.000771868 0.005188676 0.000000000
5 0 2.146517253 -0.000113530 0.003220298 0.000000000
6 1 2.146497679 -0.000019574 0.000002801 0.000000000
7 2 2.146497679 0.000000000 0.000001074 0.000000000
8 3 2.146497679 0.000000000 0.000000412 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS 2.1464976789 AFTER 8 ITERATIONS
--------------------
SPIN SZ = 0.500
S-SQUARED = 0.750
--------------------
------------
EIGENVECTORS
------------
1 2
3 4
5
-2.2253
-1.6811 -1.6389 -1.4275
-1.3467
AG AU
BU AG
BG
1 H 1 S 0.521020 0.655043
0.000000 0.490401 0.000000
2 H 2 S 0.521020 -0.655043 0.000000
0.490401 0.000000
3 H 3 S 0.133586 -0.212958 0.451368
-0.469839 0.595014
4 H 4 S 0.133586 0.212958 -0.451368
-0.469839 0.595014
5 H 5 S 0.133586 0.212958
0.451368 -0.469839 -0.595014
6 H 6 S 0.133586 -0.212958 -0.451368
-0.469839 -0.595014
MO1


MO4


6
-1.1713
AU
1 H 1 S 0.570942
2 H 2 S -0.570942
3 H 3 S 0.586689
4 H 4 S -0.586689
5 H 5 S -0.586689
6 H 6 S 0.586689
MO6

...... END OF ROHF CALCULATION ......
CPU TIME: STEP = 0.00 , TOTAL = 0.2 SECONDS ( 0.0 MIN)
WALL CLOCK TIME: STEP = 0.00 , TOTAL = 0.2 SECONDS ( 0.0 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -2.2253389866
TWO ELECTRON ENERGY = 0.0000000000
NUCLEAR REPULSION ENERGY = 4.3718366655
------------------
TOTAL ENERGY = 2.1464976789
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.0000000000
NUCLEUS-ELECTRON POTENTIAL ENERGY = -2.7062171835
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 4.3718366655
------------------
TOTAL POTENTIAL ENERGY = 1.6656194820
TOTAL KINETIC ENERGY = 0.4808781969
VIRIAL RATIO (V/T) = -3.4637034754
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -2.2253389866
BARE H ENERGY= -2.2253389866
ELECTRONIC ENERGY = -2.2253389866
KINETIC ENERGY= 0.4808781969
N-N REPULSION= 4.3718366655
TOTAL ENERGY= 2.1464976789
SIGMA PART(1+2)= -2.2253389866
(K,V1,2)= 0.4808781969 -2.7062171835 0.0000000000
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= 2.1464976789 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
1.000000
1 0.394442
2 0.394442
3 0.052779
4 0.052779
5 0.052779
6 0.052779
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H1 0.394442 0.376401
2 H1 0.394442 0.376401
3 H3 0.052779 0.061800
4 H3 0.052779 0.061800
5 H3 0.052779 0.061800
6 H3 0.052779 0.061800
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.39444 0.37640
2 H 2 S 0.39444 0.37640
3 H 3 S 0.05278 0.06180
4 H 4 S 0.05278 0.06180
5 H 5 S 0.05278 0.06180
6 H 6 S 0.05278 0.06180
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 0.2714615
2 0.0648620 0.2714615
3 0.0078848 0.0211744 0.0178451
4 0.0211744 0.0078848 0.0003153 0.0178451
5 0.0211744 0.0078848 0.0049636 0.0005958 0.0178451
6 0.0078848 0.0211744 0.0005958 0.0049636 0.0003153
6
6 0.0178451
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H1 0.394442 0.605558 0.376401 0.623599
2 H1 0.394442 0.605558 0.376401 0.623599
3 H3 0.052779 0.947221 0.061800 0.938200
4 H3 0.052779 0.947221 0.061800 0.938200
5 H3 0.052779 0.947221 0.061800 0.938200
6 H3 0.052779 0.947221 0.061800 0.938200
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.550 0.311
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H1 0.633 0.478 0.156
2 H1 0.633 0.478 0.156
3 H3 0.103 0.100 0.003
4 H3 0.103 0.100 0.003
5 H3 0.103 0.100 0.003
6 H3 0.103 0.100 0.003
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H1 1.0 0.1616790
2 H1 1.0 0.1616790
3 H3 1.0 0.0146334
4 H3 1.0 0.0146334
5 H3 1.0 0.0146334
6 H3 1.0 0.0146334
H64+, Singulett
Input
!
! H6, verzeigt,wie Bicyclobut, 4plus, Singulett
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=4 RUNTYP=ENERGY COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H6, verzweigt, wie Bicyclobut, 4+, Singulett
CNH 2
H1
H2 1 1.55
H3 1 1.38 2 87.8
H4 2 1.38 1 87.8 3 0.0 0
H5 1 1.38 2 87.8 4 -180.0 0
H6 2 1.38 1 87.8 3 180.0 0
$END
Output
-------------------
RHF SCF CALCULATION
-------------------
NUCLEAR ENERGY = 4.3718366655
MAXIT = 30 NPUNCH= 2
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F DEM=F SOSCF=F
DENSITY CONV= 1.00E-05
MEMORY REQUIRED FOR RHF STEP= 7833 WORDS.
ITER EX DEM TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0 0.388998527 0.388998527 0.114720403 0.000000000
2 1 0 0.362788352 -0.026210175 0.022573464 0.000000000
3 2 0 0.361861663 -0.000926689 0.004491858 0.000000000
4 3 0 0.361825448 -0.000036215 0.000898328 0.000000000
5 0 0 0.361824003 -0.000001445 0.000224884 0.000000000
6 1 0 0.361823943 -0.000000060 0.000000006 0.000000000
7 2 0 0.361823943 0.000000000 0.000000001 0.000000000
-----------------
DENSITY CONVERGED
-----------------
TIME TO FORM FOCK OPERATORS= 0.0 SECONDS ( 0.0 SEC/ITER)
TIME TO SOLVE SCF EQUATIONS= 0.0 SECONDS ( 0.0 SEC/ITER)
FINAL ENERGY IS 0.3618239427 AFTER 7 ITERATIONS
------------
EIGENVECTORS
------------
1 2 3 4 5
-1.7948 -1.3231 -1.2638 -1.0530 -1.0211
AG BU AU AG BG
1 H 1 S 0.445306 0.000000 0.564412 0.560052 0.000000
2 H 2 S 0.445306 0.000000 -0.564412 0.560052 0.000000
3 H 3 S 0.199584 0.451368 -0.296545 -0.445825 0.595014
4 H 4 S 0.199584 -0.451368 0.296545 -0.445825 0.595014
5 H 5 S 0.199584 0.451368 0.296545 -0.445825 -0.595014
6 H 6 S 0.199584 -0.451368 -0.296545 -0.445825 -0.595014
6
-0.7945
AU
1 H 1 S 0.660678
2 H 2 S -0.660678
3 H 3 S 0.549196
4 H 4 S -0.549196
5 H 5 S -0.549196
6 H 6 S 0.549196
...... END OF RHF CALCULATION ......
CPU TIME: STEP = 0.06 , TOTAL = 0.5 SECONDS ( 0.0 MIN)
WALL CLOCK TIME: STEP = 0.06 , TOTAL = 0.5 SECONDS ( 0.0 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -4.4304454718
TWO ELECTRON ENERGY = 0.4204327490
NUCLEAR REPULSION ENERGY = 4.3718366655
------------------
TOTAL ENERGY = 0.3618239427
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.4204327490
NUCLEUS-ELECTRON POTENTIAL ENERGY = -5.3047849537
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 4.3718366655
------------------
TOTAL POTENTIAL ENERGY = -0.5125155392
TOTAL KINETIC ENERGY = 0.8743394819
VIRIAL RATIO (V/T) = 0.5861745349
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -3.5895799741
BARE H ENERGY= -4.4304454718
ELECTRONIC ENERGY = -4.0100127229
KINETIC ENERGY= 0.8743394819
N-N REPULSION= 4.3718366655
TOTAL ENERGY= 0.3618239426
SIGMA PART(1+2)= -4.0100127229
(K,V1,2)= 0.8743394819 -5.3047849537 0.4204327489
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= 0.3618239426 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2.000000
1 0.639783
2 0.639783
3 0.180109
4 0.180109
5 0.180109
6 0.180109

----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.63978 0.61995
2 H 2 S 0.63978 0.61995
3 H 3 S 0.18011 0.19002
4 H 4 S 0.18011 0.19002
5 H 5 S 0.18011 0.19002
6 H 6 S 0.18011 0.19002
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 0.3965942
2 0.0947607 0.3965942
3 0.0201369 0.0540770 0.0796678
4 0.0540770 0.0201369 0.0014077 0.0796678
5 0.0540770 0.0201369 0.0221596 0.0026598 0.0796678
6 0.0201369 0.0540770 0.0026598 0.0221596 0.0014077
6
6 0.0796678
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H1 0.639783 0.360217 0.619952 0.380048
2 H1 0.639783 0.360217 0.619952 0.380048
3 H3 0.180109 0.819891 0.190024 0.809976
4 H3 0.180109 0.819891 0.190024 0.809976
5 H3 0.180109 0.819891 0.190024 0.809976
6 H3 0.180109 0.819891 0.190024 0.809976
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.550 0.409 1 3 2.035 0.115 1 4 1.380 0.115
1 5 1.380 0.115 1 6 2.035 0.115 2 3 1.380 0.115
2 4 2.035 0.115 2 5 2.035 0.115 2 6 1.380 0.115
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H1 0.870 0.870 0.000
2 H1 0.870 0.870 0.000
3 H3 0.328 0.328 0.000
4 H3 0.328 0.328 0.000
5 H3 0.328 0.328 0.000
6 H3 0.328 0.328 0.000
H64+, Triplett
Input
!
! H6, verzeigt,wie Bicyclobut, 4plus, Triplett
!
$CONTRL SCFTYP=ROHF MULT=3 ICHARG=4 RUNTYP=ENERGY COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
H6, verzweigt, wie Bicyclobut, 4+, Triplett
CNH 2
H1
H2 1 1.55
H3 1 1.38 2 87.8
H4 2 1.38 1 87.8 3 0.0 0
H5 1 1.38 2 87.8 4 -180.0 0
H6 2 1.38 1 87.8 3 180.0 0
$END
Output
--------------------
ROHF SCF CALCULATION
--------------------
NUCLEAR ENERGY = 4.3718366655
MAXIT = 30 NPUNCH= 2 MULT= 3
EXTRAP=T DAMP=F SHIFT=F RSTRCT=F DIIS=F SOSCF=F
DENSITY CONV= 1.00E-05
ROHF CANONICALIZATION PARAMETERS
C-C O-O V-V
ALPHA -0.5000 0.5000 1.5000
BETA 1.5000 0.5000 -0.5000
MEMORY REQUIRED FOR UHF/ROHF STEP= 8387 WORDS.
ITER EX TOTAL ENERGY E CHANGE DENSITY CHANGE DIIS ERROR
1 0 0.511032969 0.511032969 0.044294705 0.000000000
2 1 0.501477274 -0.009555694 0.018868401 0.000000000
3 2 0.499901482 -0.001575792 0.007761969 0.000000000
4 3 0.499642450 -0.000259032 0.003162398 0.000000000
5 0 0.499599893 -0.000042558 0.002159209 0.000000000
6 1 0.499591527 -0.000008366 0.000000719 0.000000000
7 2 0.499591527 0.000000000 0.000000292 0.000000000
-----------------
DENSITY CONVERGED
-----------------
FINAL ENERGY IS 0.4995915268 AFTER 7 ITERATIONS
--------------------
SPIN SZ = 1.000
S-SQUARED = 2.000
--------------------
------------
EIGENVECTORS
------------
1 2 3 4 5
-1.9604 -1.6557 -1.3971 -1.2055 -1.0978
AG BU AU AG BG
1 H 1 S 0.450655 0.000000 0.628284 0.555756 0.000000
2 H 2 S 0.450655 0.000000 -0.628284 0.555756 0.000000
3 H 3 S 0.195300 0.451368 -0.239531 -0.447718 0.595014
4 H 4 S 0.195300 -0.451368 0.239531 -0.447718 0.595014
5 H 5 S 0.195300 0.451368 0.239531 -0.447718 -0.595014
6 H 6 S 0.195300 -0.451368 -0.239531 -0.447718 -0.595014
6
-0.8926
AU
1 H 1 S 0.600263
2 H 2 S -0.600263
3 H 3 S 0.576351
4 H 4 S -0.576351
5 H 5 S -0.576351
6 H 6 S 0.576351
...... END OF ROHF CALCULATION ......
CPU TIME: STEP = 0.05 , TOTAL = 0.2 SECONDS ( 0.0 MIN)
WALL CLOCK TIME: STEP = 0.05 , TOTAL = 0.2 SECONDS ( 0.0 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
-------------------------------
properties for the ROHF density
-------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -4.1284081042
TWO ELECTRON ENERGY = 0.2561629654
NUCLEAR REPULSION ENERGY = 4.3718366655
------------------
TOTAL ENERGY = 0.4995915268
ELECTRON-ELECTRON POTENTIAL ENERGY = 0.2561629654
NUCLEUS-ELECTRON POTENTIAL ENERGY = -5.2162991176
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 4.3718366655
------------------
TOTAL POTENTIAL ENERGY = -0.5882994866
TOTAL KINETIC ENERGY = 1.0878910134
VIRIAL RATIO (V/T) = 0.5407706097
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -3.6160822493
BARE H ENERGY= -4.1284081042
ELECTRONIC ENERGY = -3.8722451767
KINETIC ENERGY= 1.0878910134
N-N REPULSION= 4.3718366655
TOTAL ENERGY= 0.4995914888
SIGMA PART(1+2)= -3.8722451767
(K,V1,2)= 1.0878910134 -5.2162991176 0.2561629274
PI PART(1+2)= 0.0000000000
(K,V1,2)= 0.0000000000 0.0000000000 0.0000000000
SIGMA SKELETON, ERROR= 0.4995914888 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2
1.000000 1.000000
1 0.325109 0.000000
2 0.325109 0.000000
3 0.087445 0.250000
4 0.087445 0.250000
5 0.087445 0.250000
6 0.087445 0.250000

ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 H1 0.325109 0.314619
2 H1 0.325109 0.314619
3 H3 0.337445 0.342690
4 H3 0.337445 0.342690
5 H3 0.337445 0.342690
6 H3 0.337445 0.342690
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 H 1 S 0.32511 0.31462
2 H 2 S 0.32511 0.31462
3 H 3 S 0.33745 0.34269
4 H 4 S 0.33745 0.34269
5 H 5 S 0.33745 0.34269
6 H 6 S 0.33745 0.34269
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 0.2030903
2 0.0485256 0.2030903
3 0.0099707 0.0267760 0.2418754
4 0.0267760 0.0099707 -0.0029259 0.2418754
5 0.0267760 0.0099707 0.0672778 -0.0055285 0.2418754
6 0.0099707 0.0267760 -0.0055285 0.0672778 -0.0029259
6
6 0.2418754
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 H1 0.325109 0.674891 0.314619 0.685381
2 H1 0.325109 0.674891 0.314619 0.685381
3 H3 0.337445 0.662555 0.342690 0.657310
4 H3 0.337445 0.662555 0.342690 0.657310
5 H3 0.337445 0.662555 0.342690 0.657310
6 H3 0.337445 0.662555 0.342690 0.657310
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.550 0.211 1 3 2.035 0.057 1 4 1.380 0.057
1 5 1.380 0.057 1 6 2.035 0.057 2 3 1.380 0.057
2 4 2.035 0.057 2 5 2.035 0.057 2 6 1.380 0.057
3 4 3.113 0.053 3 5 1.444 0.228 3 6 2.758 0.053
4 5 2.758 0.053 4 6 1.444 0.228 5 6 3.113 0.053
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 H1 0.545 0.439 0.106
2 H1 0.545 0.439 0.106
3 H3 0.561 0.447 0.114
4 H3 0.561 0.447 0.114
5 H3 0.561 0.447 0.114
6 H3 0.561 0.447 0.114
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 H1 1.0 0.1251288
2 H1 1.0 0.1251288
3 H3 1.0 0.1439498
4 H3 1.0 0.1439498
5 H3 1.0 0.1439498
6 H3 1.0 0.1439498